")
Back to Journals » Biologics: Targets and Therapy » Volume 16
A Review on Inflammatory Bowel Diseases: Recent Molecular Pathophysiology Advances
Authors Abdulla M , Mohammed N
Received 25 June 2022
Accepted for publication 27 August 2022
Published 12 September 2022 Volume 2022:16 Pages 129—140
DOI https://doi.org/10.2147/BTT.S380027
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Doris Benbrook
Maheeba Abdulla,1 Nafeesa Mohammed2
1Internal Medicine Department, Ibn AlNafees Hospital, Arabian Gulf University, Manama, Bahrain; 2Salmaniya Medical Complex, Manama, Bahrain
Correspondence: Maheeba Abdulla, Consultant Gastroenterologist, Internal Medicine Department, Ibn AlNafees Hospital, Arabian Gulf University, Manama, Bahrain, Email [email protected]
Abstract: Inflammatory bowel diseases are considered immune disorders with a complex genetic architecture involving constantly changing endogenous and exogenous factors. The rapid evolution of genomic technologies and the emergence of newly discovered molecular actors are compelling the research community to reevaluate the knowledge and molecular processes. The human intestinal tract contains intestinal human microbiota consisting of commensal, pathogenic, and symbiotic strains leading to immune responses that can contribute and lead to both systemic and intestinal disorders including IBD. In this review, we attempted to highlight some updates of the new IBD features related to genomics, microbiota, new emerging therapies and some major established IBD risk factors.
Keywords: inflammatory bowel disease, immune disorders, genomics, ulcerative colitis, Crohn’s disease
Introduction
Research in the inflammatory bowel disease (IBD) field is considered one of the most relevant challenges of modern gastroenterology. IBD is becoming more well recognized as a complex condition that can be brought on or made worse by a variety of causes, including genetic predisposition, environmental variables including smoking and diet, varying immune responses, and changes in the microbiota present in the intestine1 and IBD prevalence is higher in developed countries and is estimated to more than 6 million patients worldwide.2 An emergence of IBD cases is observed in developing countries; newly industrialized countries are experiencing an acceleration of IBD incidence in India, South Korea, China and North Africa and Middle East explained by gradual lifestyle westernization, essentially consisting in the change of eating habits, leading to an increase of IBD incidence. Western regions are undergoing the compounding prevalence stage. The evolution of IBD is related to industrial expansion specifically in North America and Western Europe with a fundamental societal shift toward urbanization, agriculture, manufacturing, and transportation. A number of studies have been published that note its effect on patients’ health wherein they suffered from numerous digestive disorders that are non-infectious and formally named ulcerative colitis.3 IBD is considered a major problem in medicine, due to the high costs of treatment, frequent disability in youngsters and active adults, and the complexity of social rehabilitation of patients.4 It also has a substantial impact on the quality of life of both patients and their families as it requires lifelong modifications to behavior, lifestyle, and dietary habits.5
IBD is represented by two pathologies: ulcerative colitis (UC) and Crohn’s disease (CD). The phenotypic spectrum of each one differs, with gastrointestinal and extra-intestinal signs as its hallmarks.6 Nevertheless they have several clinical similarities, as they lead to ulceration. Clinical manifestations present as periods of relapses interspersed with periods of remissions. In contrast; there are some specific differences between UC and CD. In fact, CD affects the whole digestive system and is typified by a transmural inflammation with skip lesions occurring from the mouth to anus, but characteristically involving the terminal ileum,119 whereas UC is characterized by an initial inflammatory process in the rectum which progressively spreads to the totality of the colon. Macroscopically, CD affects the intestinal epithelium by causing transmural inflammation, successively degrading mucosa, submucosa, musculosa externa and serosa adventive, whereas UC affects only mucosa and submucosa.7
Additionally, some gastrointestinal illnesses can closely resemble IBD, which can delay diagnosis and make choosing a course of treatment more difficult. The challenges are exacerbated by limitations on endoscopic capability and a lack of skilled diagnostic pathologists.8 Over decades, significant research progress has been accomplished in the field of IBD in order to unravel the IBD pathogenesis. Genetic, genomics and molecular biology advances have provided the most relevant data in terms of genes involved, gene-gene and gene-environment interactions, molecular pathways, and immunity and microbiota factors. In this review, we updated some recent major breakthroughs in the aforementioned areas.
Genetic and Genomic Advances
Research has shown the existence of a genetic component in the onset of IBD. Indeed, epidemiological studies have demonstrated a high genetic contribution (λs ~ 15–42) for CD,6–10 whereas for UC the genetic contribution was lower (λs ~ 7–17).9,10 To date, GWAS has identified nearly 250 loci associated with IBD10–14 For CD, genotyping of the NOD2 gene has demonstrated its involvement in disease-onset.
According to early research, mutations in NOD2 gene mutations are linked to an increased risk of CD onset of 40%. The NOD2 gene codes for an intra-cytoplasmic protein which has two domains that play roles in the recognition of proteins involved in apoptosis, in the activation of NF-Kb pathways. NOD2 is expressed in Paneth cells situated in the ileum that has a lesser degree of epithelial cells compared to the lungs, oral cavity, or intestines. NOD2 also has bacterial components like LPS, hormonal vitamin D, short-chain fatty acids like butyrate, and pro-inflammatory cytokines like TNF-alpha. It is a part of 110kDa cytosolic protein that is part of the NLRC subfamily. It has two CARD domains and has a human chromosome 16p21. The central NOD domain plays a role in protein oligomerization. The C-terminal domain, LRR, is involved in the recognition of bacterial components such as muramyl dipeptide, present in the peptidoglycan, which is an essential element of the bacterial cell wall.NOD2 is expressed by immune cells such as macrophages, lymphocytes, dendritic cells but also by Paneth cells.15
Research on NOD2 gene involvement in CD susceptibility has been performed since the beginning of the XXI century on different European populations due to a continual incidence increase of the pathology in the Western world.16–21 It was assumed that the lower prevalence of IBD in Asia was due to the lower presence of genes susceptible to IBD in Asians, but recently a rapid increase in the incidence and prevalence of IBD has been noted in the Asian population22,23 leading to an extended research on NOD2 variant prevalence in Asia. Also, numerous, recent case-control studies have been published for the Arab population.24–27 It has also been demonstrated that the NOD2gene serves as an independent prognostic factor for clinical CD behavior.28 In addition, NOD2 haplotypic contribution has been evidenced for pediatric CD29 in which population attributable risk and total heritability of NOD2 gene was 32.9% and 3.4% respectively.30
MiRNA regulation of the NOD2 signaling pathway was reported by Chuang et al: overexpressed miR-495, miR-192, miR-671, and miR-512 had led to an abolishment of NOD2 mRNA expression, muramyl dipeptide-mediated NF-κB activation, and mRNA expressions of interleukin-8 and CXCL3 in HCT116 cells. Also, a NOD 2 3′-UTR SNP (rs3135500) reduced miR-192 effects on NOD2 gene expression.31
Most recent studies have found significant evidence in favor of 21 new loci, located within NOD2, LRRK1, WHAMM, DENND3, C5, TPL2, TNFSF1 MHC, and MST1 3p21 genes, which are important in the pathogenesis of IBD.10–14 Fachal et al, have identified novel genome-wide significant signals in joint effort with the International IBD Genetics Consortium (32 loci associated with CD, 36 with UC, 106 with IBD). Among them, six newly identified loci were imputed to monogenic syndromes that include colitis: CARMIL2, DOCK8, G6PC3, HPS4, NCF1, PIK3CD.32
In addition, recent studies34 of the mechanisms of interaction of the host genotype with the environment in the formation of an individual CD phenotype have shown the important role of epigenetics as a mediating link in the realization of some environmental effects, genetic predispositions, and intestinal microbiota in the pathogenesis of the formation and persistence of chronic inflammation in the intestine. Genetic susceptibility plays an important role in the development of IBD as a number of large genomic studies show that there are more than 200 risk alleles that can trigger a spectrum of disease phenotypes related to IBD. It is important to identify and define the heritability of IBD as host genetics can influence the gut microbiota leading to regulated and balanced immune responses with coordinated microbial composition. Various interactions studied in multiple genomic studies show that in IBD patients, the nature of their microbiota changes, leading to its inflammation and alteration.33,35 In order to provide more tailored treatment for CD, it is now widely acknowledged that the investigation of epigenetic alterations, such as DNA methylation or histone modification, is very helpful in identifying new biomarkers or targets for pharmacological therapy.36 Examples of clinical biomarkers for diagnostic, prognostic, and therapeutic reasons include epigenetic changes in colorectal cancer, allergies and asthma, and cardiovascular disease.37
The importance of innate host immune response to bacteria is demonstrated by genetic discoveries related to participation in the pathogenesis of IBD, the process of intracellular recognition of bacteria and the mechanisms of intracellular processing of bacteria, primarily autophagy.38 An intracellular catabolic process called autophagy is necessary for a number of cellular responses. The dysregulation or interruption of autophagy may be related to IBD since it plays a part in preserving biological homeostasis under stress.39 Recently, it has been demonstrated that autophagy-related genes (ATGs), including optineurin (OPTN), transcription factor EB (TFEB), and leucine-rich repeat kinase (LRRK), are linked to an increased risk of developing colitis, indicating that these genes are crucial for maintaining colonic immune homeostasis.40 It is now evident that autophagy activation helps to reduce excessive inflammatory reactions.41 Containing the nucleotide-binding domain of oligomerization protein 2(NOD2), located at locus 16q12, plays an important role in the immune system by controlling the commensal microbiota in the intestine.15
NOD2 belongs to the leucine repeat-rich family of cytoplasmic proteins that can recognize various bacteria, acting as intracellular sensors for bacterial peptidoglycans. This protein binds to the muramyldipeptide of gram-positive and gram-negative bacteria, leading to the activation of signaling pathways mediated by the Nuclear Factor (NFkB). NFkB is the main transcriptional regulator of proinflammatory cytokines, including tumor necrosis factor α (TNF-α), which plays an important role in the development of inflammation in CD.42
The existence of monogenic IBD, which is brought on by a single gene deficiency, is one of the most crucial pieces of evidence. Over the past few years, translational research into monogenic IBD has advanced quickly, and there have been multiple reports of unique single gene mutations causing IBD.43 The number of monogenic IBD disorders is increasing, with genes divided into six groups based on the biologic mechanism. These groups include epithelial barrier defects, T- and B-cell defects, hyper- and autoinflammatory disorders, phagocytic defects, immunoregulation, including IL-10 signaling defects, and other.44 Monogenic IBD frequently exhibits partial autosomal dominant and autosomal recessive IBD phenotypic expression. For instance, IBD-like phenotypes are seen in 20–30% of patients with XIAP deficiency or GSD-1b caused by SLC374A4,45 and in 4–9% of patients with Wiskott Aldrich Syndrome,46 and it is hypothesized that unrelated modifier genes, epigenetic alterations, or environmental variables cause incomplete expressivity.47 In contrast to monogenic IBD, the effects of genetics on classical IBD (polygenic IBD) are also considerable.48 Our knowledge of the genetic makeup and underlying mechanisms behind IBD has significantly increased as a result of genome-wide association studies (GWAS), which have discovered roughly 240 disease loci associated with the classic adult-onset form of the disease.49 Similar to this, prevalent IBD variations connected to childhood-onset IBD have been researched; the majority of these variants are connected to adult-onset IBD.50
The Role of the Microbiome in the Pathogenesis of IBD
Under IBD condition, the influence of combined endogenous (genetic) and exogenous (genetic, microbial, dietary, stress) factors can lead to intestinal barrier dysfunction by increasing its permeability, which promotes translocation of microorganisms and products of microbial origin from the intestinal lumen into the mucous layer and intestinal epithelium, subsequently leading to an activation of immune cells (Th1/Th2/Th17), a Th17 /Treg imbalance and cytokine production with consequent development of chronic inflammation. In turn, the chronicity of the inflammation worsens the already existing violations of the barrier function.51,52
IBD can be considered a kind of “polymicrobial” disease, in which the altered intestinal microbiota – dysbiosis has a major role.53,54
The most important pro-inflammatory cytokines in IBD are Tumor Necrosis Factor α (TNF-α), Interleukins (IL) IL-6 and IL-23. During CD, the development of chronic inflammation is mediated by Th1 and Th17 cells involving IFNγ, IL-6, IL-12, TNF-α (Th1) and IL-17, IL21, IL-22 (Th17), respectively, whereas in UC, inflammation is mediated by Th2 and Th17 cells involving IL-4, IL-5, IL-6, IL-13, IL15, IL-33, TNF-α (Th2) and IL-17, IL-21, IL-22 (Th17).55,56
IL33 can play a key role in the interaction with the intestinal microbiota in order to maintain homeostasis and ensure protective functions of the intestinal barrier, as well as in the regulation of adaptive immunity and Th17/Treg balance. At the same time, the IL-33/ST2 signaling axis (through the induction of an IL-4-dependent immune response) is involved in the pathogenesis of IBD, in the active phase of which the soluble stimulating growth factor expressed by gene 2 (sST2) is secreted by proinflammatory intestinal T cells, and the number of protective ST2-expressing Treg level decrease.57,58
The mechanism of possible participation of the intestinal microbiota in the development and maintenance of the inflammatory process in the intestine can be represented as follows: it is assumed that in the conditions of microbial homeostasis, symbiotic microorganisms have a predominantly anti-inflammatory effect, by suppressing pathobionts characterized by a potential colitogenic effect by inducing an immune response involving intestinal regulatory T cells (Treg), anti-inflammatory interleukin IL-10 and restoring the REG3i and REG3G levels. In IBD, a combination of genetic (NOD2/CARD15 gene mutations, ATG16L1 autophagy gene and IL-23 receptor gene) and environmental factors cause both a violation of the barrier function of the intestinal mucosa and damage to the structure of the microbiota (intestinal dysbiosis). A decrease in the number of “protective” symbiotic bacteria and/or an increase in the number of pathobionts, characteristic of the state of dysbiosis, support and worsen the inflammatory process.51,59
Evidence points to abnormalities in innate and adaptive immune responses against intestinal microbiota, harmful antigens, or extrinsic pathogens that may have crossed the intestinal barrier as playing a significant role in the inflammatory process associated with the disease in those who are genetically susceptible.60 Innate lymphoid cells, innate immune response (macrophages, neutrophils, and dendritic cells), and adaptive immune response (T and B cells) cells, as well as various cytokine and chemokine types that are released by these cells, are all implicated in the pathogenesis of IBD.61 Previous research has shown that Th1-related cytokines, such as tumor necrosis factor (TNF), interferon (IFN)-, and interleukin (IL)-12, as well as Th17-associated cytokines, such as IL-17A, IL-21, and IL-23, are markedly increased in the inflamed mucosa of CD patients, whereas the cytokine profiles in the inflamed areas.62,63
Given the significance of Tregs in controlling immunological responses, it has been proposed that abnormalities in Tregs and their mediators play a key role in the pathogenesis of IBD. Despite the fact that there are fewer Tregs in the peripheral blood during active IBD compared to quiescent IBD and controls,64 their anti-inflammatory capability is still present as evidenced by their capacity to inhibit effector T-cell proliferation in vitro.65 It is possible to infer that the immunopathogenesis of IBD is significantly influenced by the ratio of effector T-cells to Tregs rather than the absolute quantity of Tregs.
An increase in the number of pro-inflammatory microorganisms may contribute to the activation of pro-inflammatory T-cells (Th17), causing, in genetically susceptible people, a Th17-mediated autoimmune response. In turn, a decrease in the number of anti-inflammatory microorganisms can lead to underdevelopment of a subpopulation of key immunoregulatory cells (Treg). An imbalance between Th17 and Treg will eventually lead to the development of autoimmune inflammation.66
Another possible mechanism of microbiota participation in the modulation of inflammation in the intestine may be its interaction with Nod-like receptors. Caspase-1 and other pro-inflammatory cytokines like IL-18, and IL-1β are activated by NOD-like receptors. It can lead to inflammatory cell death, secretion and processing of inflammatory cytokines, colonization of gastrointestinal bacterial pathogens, and shaping of gut microbiota that plays a critical role in IBD.67
In addition, a possible role was demonstrated in an experimental study on CARD9-associated disorders of tryptophan microbial metabolism in the pathogenesis of IBD mediated by aryl-hydrocarbon receptors (AhR).68
Role of Dysbiosis in IBD
Although a precise description of a healthy gut microbiome has not yet been established, dysbiosis is typically referred to as a disrupted equilibrium between the microbiota and its host.69 As a result, commensal microbiota are essential for the early development of the immune system and microbes, and their byproducts regulate immunological responses by inducing immune cells, communication pathways, and inflammatory mediators.70 In contrast, environmental factors, such as dietary elements, gastrointestinal infections, drugs, psychological stress, and smoking, cause dysbiosis-associated mucosal immune dysfunction in genetically predisposed individuals, which distinguish IBD. The mucosal immune response becomes dysfunctional under chronic dysbiotic circumstances, which are marked by a rise in aggressive bacterial strains and a decrease in regulatory species (Figure 1). Gut dysbiosis is likely to prolong mucosal inflammation and potentially result in IBD when combined with impaired intestinal barrier function.71 IBD risk factors include altered endoplasmic reticulum stress responses, a thinner mucus layer, and impaired physical epithelial barrier function (through mutations in MUC19, ITLN1, FUT2, and XBP1).72 Currently, it is thought that exposure to gut antigens, such as microbiome components, in genetically predisposed people causes an incorrect immune response that leads to the development of human IBD.73 Although it has been suggested that changes in the gut microbiome are crucial for IBD pathogenesis, it is still unclear how these changes take place and if dysbiosis is the disease’s primary cause or a frequent complication.74
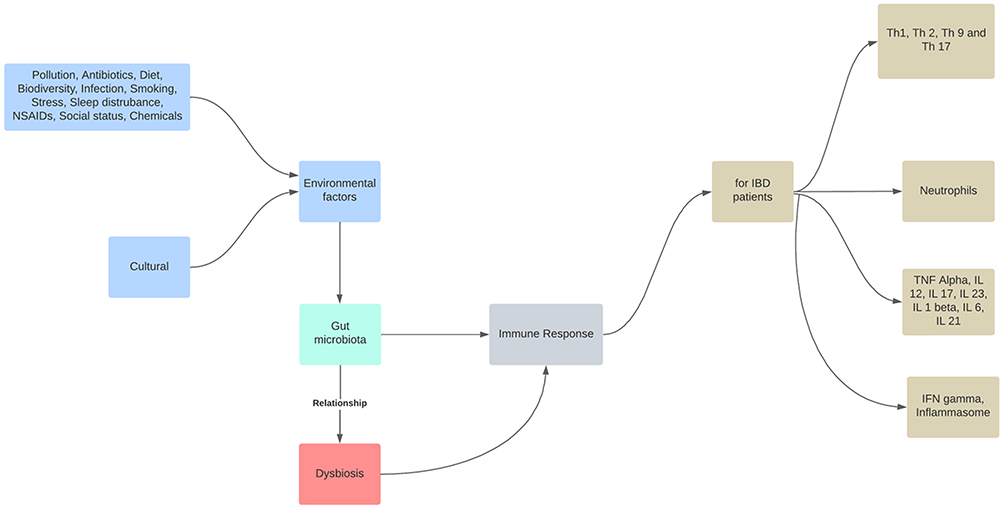 |
Figure 1 The role of intestinal dysbiosis plays in the development of IBD. The gut microbiota are a reflection of the interaction between the host’s genetics and dynamic exposure to countless stimuli from the exposome. Notes: Role of dysbiosis in IBD. Adapted from Santana PT, Rosas SLB, Ribeiro BE, Marinho Y, de Souza HSP. Dysbiosis in inflammatory bowel disease: pathogenic role and potential therapeutic targets. Int J Mol Sci. 2022;23(7):3464. doi:10.3390/ijms23073464.75 |
IBD Impacts on Pregnancy and Offspring
Offspring born to a parent with CD have a 2 to 3% risk of being further affected by the disease compared to 0.5 to 1% for UC. On the other hand, if both parents have IBD, the risk increases to 30%.76
As IBD has been previously shown to be a polygenic disease involving a large number of low penetrance genes, modifiable intrinsic factors play a large role in the development of IBD especially in the microbiome. It would be useful and topical to highlight the advances made on the research front to elucidate the influence of maternal microbiota during pregnancy on that of the fetus and the newborn.77
According to Zhou et al, during gestation, the intestinal microbiota in the first trimester is similar to the microbiota during non-pregnant state, with similar proportions of Bacteroidetes and Firmicutes reported by.78 During the second trimester, the most predominant abundant phyla in the microbial community structure were Firmicutes, Bacteroidetes, Actinobacteria, Tenericutes, and Proteobacteria, and the abundances of Bifidobacteriaceae, and Enterobacteriaceae were relatively increased.79
During the third trimester, the maternal gut microbiota changes, with a decrease of alpha diversity, butyrate-producing bacteria, such as Faecalibacterium, and the increase of beta diversity, Actinobacteria, Bifidobacterium, Enterobacteriaceae, Streptococcus, and Proteobacteria.78,80,81
Kennedy et al demonstrated that fetal gut colonization of healthy term infants does not occur before birth and that microbial profiles of neonatal meconium reflect populations acquired during and after birth.77
Torres et al demonstrated the existence of an alteration of the bacterial composition in the microbiota of pregnant IBD patients and that the bacterial diversity evolves with an enrichment of Gammaproteo bacteria and depletion of Bacteroidetes, and infants born to mothers with IBD showed lower diversity and altered bacterial gut composition in up to at least 3 months of life. Adaptive immune system is influenced by the induced alteration of microbiota in germ-free mice used as an animal model, suggesting that offspring’s immune system may be impacted by microbial factors in maternal IBD.82
Targeting perturbations of the microbiome in pregnant women with IBD or neonates could contribute to maintaining a healthy microbiome in offspring, and primary interventions at this stage could be established in order to prevent the development of IBD later in life83,84 as was further aimed by the MELODY trial which had investigated the effect of bacterial manipulation through diet during the third trimester of pregnancy on microbiome composition improvement in the gut of CD patients in order to lead to the development of a healthier microbiome in offspring.85
Gueimonde et al, reported that probiotics taken by pregnant women can pass through their intestinal microbiota to the fetus through the placenta, and consequently affect fetal intestinal microbiota.86
Stinson et al reported that infant intestinal microbiota could originate from amniotic fluid.87 Also, the constant ingestion of amniotic fluid and its microorganisms by the fetus could be important in the modulation of infant intestinal microbiota due to the contact of the ingested amniotic fluid with fetal intestine. Developmental symbiosis and plasticity of the intestines play a critical role in the development of IBD. It is related to genetic variability, facilitated by evolutionary transitions. Novel phenotypes are generated that can facilitate evolutionary transitions and the development of genetic accommodation and plasticity of the gut in IBD.88
He et al analysis of 39 pairs of infant meconium samples and amniotic fluid, and faeces, vaginal fluid, and saliva from mothers demonstrated that the microbes in meconium came from multiple sources but mainly from amniotic fluid microbiota.89
Galazzo et al reported that vaginally delivered infants obtain a significant enrichment of Bacteroides90.
Meyer et al demonstrated IBD women’s pregnancies had increased risks of prematurity, small for gestational age, and cesarean section, especially among active IBD patients. Disease activity was decreased during pregnancy in women with CD, but was unchanged in women with UC.91
Hill et al demonstrated that vaginal microbiota presented a prevalence of Mollicutes, and has been associated with preterm delivery.92
Promising Therapeutic Strategies
By using medication, such as aminosalicylates, corticosteroids, immunomodulators, and biologics, together with additional general therapies and/or surgical resection if necessary, traditional treatments try to control symptoms. However, a sizable portion of patients do not respond to current treatments or experience a loss of response, necessitating the development of novel therapeutic approaches.93
Epigenetics can be considered in the context of elaboration of future therapy such as MSCs, MSC-driven exosomal microRNAs, and MSC-based drug delivery systems that have been demonstrated to affect immune system modulation in experimental animal models of IBD. However, further clinical trials are still needed.94
JAK inhibitors (as Tofacitinib) are also a promising path in the treatment of IBD, providing an advantage to lacking immunogenicity and showing efficacy in the treatment of moderate to severe UC.95
Promising results have been shown in both induction and maintenance therapy with IL-23 inhibitors in CD and UC. Clinical and molecular predictors of response should be explored to target the patients who are most likely to benefit from these therapies.96
A promising new idea for the treatment of IBD is oligonucleotide-based treatments. The biochemical actions of oligonucleotides can activate cellular targets for immunomodulation by mimicking bacterial DNA or by inhibiting the translation of mRNA transcripts of pro-inflammatory molecules.97
Numerous physiological functions, such as intestinal homeostasis, gastrointestinal motility regulation, visceral sensation, or immunomodulation of inflammation in inflammatory bowel disease (IBD), are modulated by the endocannabinoid system (ECS). It is made up of cannabinoid receptors (CB1 and CB2) and the enzymes responsible for their synthesis and degradation. The manipulation of these enzymes through system agonists and antagonist shows a potential therapeutic role for ECS in IBD.98
A possible treatment for UC is berberine. However, little is known about the pathophysiology of UC and the therapeutic targets of berberine. The proteins and pathways linked to the onset of colitis and its resolution following berberine administration were characterized using iTRAQ-based proteomics.99 Additionally, patient education improves the effectiveness of IBD treatment in some ways.94
Environmental Risk Factors
There are clearly identified susceptibility genes involved in the genesis of IBD. Nevertheless, the genetic factor does not fully explain the occurrence of the disease.
Smoking is considered a factor favoring the development of CD and, conversely, a protective element against the occurrence of UC. Studies focusing on clinical and sociodemographic characteristics of IBD patients showed that 80% of patients were former smokers with predominance in CD patients.100
According to Spekhorst et al in 2017, 44% of patients with CD had reported tobacco consumption compared to 18% for cases of UC. Patients afflicted with IBD are increasing every year, increasing the burden on society with symptoms related to abdominal discomfort, weight loss, fatigue, diarrhea, rectal bleeding, and other mild symptoms. Therapeutic intervention with expensive medical and surgical maneuvers is required to reduce the impact of IBD on people across the globe.101
Conversely, a Chinese -Indian -American study comparing the prevalence of smoking at the time of diagnosis of IBD among different populations showed that in China, 13.7% of UC patients were active smokers compared to 11.5% for CD patients. In India, active smoking was statistically more representative in the CD subgroup (8%) than in the UC subgroup (4.6%). For the United States, the prevalence of smokers within the UC subgroup was 25.2% compared to 19.8% for CD.102
In the evolution of the disease, various studies have highlighted an increase in flare-ups and complications in smokers, namely abscesses and fistulas, compared to non-smokers. The analysis of the therapeutic management in smoking/non-smoking patients revealed a difference between the two groups. Indeed, it is possible to notice an earlier use of immunosuppressants and corticosteroids in the smokers group. Similarly, smoking increases the risk of postoperative relapse in CD. On the other hand, weaning reduces relapse risk in CD.103,104
The smoking effect on CD could be explained by different mechanisms. It was suggested that tobacco exerts an immunomodulatory action by causing a decrease in IL-8, IL-1B and TNF alpha. Secondly, smoking causes an increase in carbon monoxide and pro-coagulant activity, which accentuates the alterations of the mucosa in particular because of ischemia. This may explain the increase in complications such as fistulas in smoking patients.105
Another hypothesis involves the mechanism of free radicals. Tobacco increases lipid peroxidation, which leads to the production of free radicals, which are harmful to the integrity of the intestinal mucosa. Finally, tobacco also has the effect of altering intestinal permeability and motility.106
It is relatively complex to answer the question about the existence of a link between eating habits and IBD. Initially, it is necessary to distinguish between the impact of diet on the onset of the disease and the impact on the evolution and on the severity of the flare-up phases. Studies have shown the beneficial effect of an anti-inflammatory diet that could reduce the frequency of IBD.107–109
Nevertheless, a diet rich in sugary products and bad quality fats, such as trans fatty acids and excess omega 6, as well as a diet low in fiber could be a risk factor. In addition, the excessive industrialization of food is also an important point because everything is washed and sometimes bleached like vegetables in sachets or even pasteurized dairy products.110
Also, the consumption of food microparticles is constantly increasing in developed countries. A study carried out on titanium dioxide, a nanoparticle used as a food additive under the name E171, showed that after ingestion, this additive can cross the intestinal barrier and end up in the blood and subsequently in the liver and further alter the immune system, induce DNA damage, and tumor promotion.111
A study, carried out on a cohort of 194,700 subjects showed that regular physical activity was associated with a reduced risk of developing Crohn’s disease. According to the results, women with physical activity equivalent to 9 hours of walking per week reduce their risk of developing CD by 44%. Indeed, the incidence of the disease for women in the “inactive” group was 11 cases per 100,000 people, while the incidence dropped to 7 cases per 100,000 people in the “active” group. It is important to note that before establishing the results, the researchers took into account other risk factors for the disease such as tobacco and BMI. On the other hand, the results of this same study do not show the protective role of physical activity for UC.112
According to a retrospective study, including approximately 220 patients with IBD, BMI had an impact on the disease. In this study, patients were divided into two groups at the time of diagnosis. The first group had a BMI superior to 25kg/m2and the second group included patients with a BMI inferior to 25kg/m2. According to the results, IBD occurring in patients with a BMI superior to 25 kg/m2 appeared at a later age and the disease seemed less severe. Indeed, during diagnosis, the patients from the first group had a better-preserved general condition, hospitalizations were less frequent and systemic corticosteroid therapy was also less frequent. In addition, the most serious forms of UC, namely the pancolitis forms, were fewer. Similarly for CD, stenosing and penetrating forms were less frequent.113
Comparing the serum vitamin D value, women with lower serum vitamin D levels have a higher risk of triggering CD. In terms of relapses, vitamin D also seems to have an impact on the course of the disease. Indeed, a study carried out on nearly 100 adults with CD receiving vitamin D3 compared to controls not supplemented with the vitamin. The results prove that the relapse rate, after one year, is lower in the group that received the treatment with 0-13% relapse for patients on vitamin and 29% for patients on placebo. This risk factor is linked to sun exposure, which may possibly help to explain the classically admitted North-South gradient reported in IBD epidemiology early studies.114,115
In the context of UC, several studies highlight the link between IBD onset and appendectomy and have demonstrated the protective factor of the operation against UC.116 On the other hand, for CD, appendectomy does not seem to be a protective factor and could even increase the risk. Indeed, acute appendicitis is often an entry form of the disease. Therefore, appendectomy may be a warning sign of undiagnosed CD.117
Conclusion and Future Perspectives
Our literature review has highlighted recent progress made in various IBD related factors. During the COVID-19 pandemic, great amounts of epidemiological data were obtained all over the world. IBD is a multifaceted and complex gastrointestinal tract disorder with increased incidence and morbidity. With advancements in both endoscopy and endomicroscopy imaging modalities, high-definition metagenomics, transcriptomics, proteomics, and other powerful tools can unravel mechanistic insights and diagnosing capabilities to address the clinical needs in patients suffering from IBD. In the current review, we emphasize how research efforts on important pathways related to innate immunity, autophagy, lymphocyte differentiation, and chemotaxis have helped us better understand the etiology of IBD. Promising candidates for patient classification and therapeutic targeting include a number of these emerging genetic markers and cellular pathways while the term “dysbiosis”, which refers to a change in the makeup and function of the gut microbiota in IBD, has recently been coined by researchers using next-generation sequencing technology. According to clinical and experimental findings, dysbiosis may be a key factor in the development of IBD. There are possibilities and obstacles as we work toward individualized and precise therapy. The indications, contraindications, and evidence-based medicine of various medications and therapies should be thoroughly understood by doctors in order to create tailored treatment plans based on a thorough evaluation of the patient. The course of treatment needs to be adaptable and modified based on the patient’s reaction and there are different environmental factors such as smoking, diet, urbanization and physical activity which have a great impact on IBD.
In order to treat the massive data obtained, new algorithms and COVID –IBD data analysis methods should be elaborated by IA, ML and DL methods.118
Funding
This study received no specific funding.
Disclosure
The authors declare that there are no conflicts of interest regarding the publication of this paper.
References
1. Kaplan GG. The global burden of IBD: from 2015 to 2025. Nature Rev. 2015;12:720–727. doi:10.1038/nrgastro.2015.150
2. Silangcruz K, Nishimura Y, Czech T, Kimura N. Impact of the world inflammatory bowel disease day and crohn’s and colitis awareness week on population interest between 2016 and 2020: google trends analysis. JMIR Infodemiol. 2021;1(1):e32856. doi:10.2196/32856
3. Kaplan GG, Windsor JW. The four epidemiological stages in the global evolution of inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2021;18(1):56–66. doi:10.1038/s41575-020-00360-x
4. Shafer A, Shaffer S, Witt J, Nugent Z, Bernstein CN. IBD Disability Index Is Associated With Both Direct and Indirect Costs of Inflammatory Bowel Disease. Inflamm Bowel Dis. 2021. doi:10.1093/ibd/izab248
5. Principi M, Losurdo G, Iannone A, et al. Differences in dietary habits between patients with inflammatory bowel disease in clinical remission and a healthy population. Ann Gastroenterol. 2018;31(4):469–473. doi:10.20524/aog.2018.0273
6. Roncoroni L, Gori R, Elli L, et al. Nutrition in patients with inflammatory bowel diseases: a narrative review. Nutrients. 2022;14(4):751. doi:10.3390/nu14040751
7. Abraham C, Cho JH. Mechanisms of disease. N Engl J Med. 2009;361:2066–2078. doi:10.1056/NEJMra0804647
8. Watermeyer G, Awuku Y, Fredericks E, et al. Challenges in the management of inflammatory bowel disease in sub-Saharan Africa. Lancet. 2022;22:S2468–S1253. doi:10.1016/S2468-1253(22)00048-6
9. Meucci G, Vecchi M, Torgano G, et al. Familial aggregation of inflammatory bowel disease in northern Italy: a multicenter study. Gastroenterology. 1992;103(2):514–519. doi:10.1016/0016-5085(92)90841-l
10. Orholm M, Munkholm P, Langholz E, et al. Familial occurrence of inflammatory bowel disease. N Eng J Med. 1991;324(2):84–88. doi:10.1056/NEJM199101103240203
11. Ben-Yosef N, Matthew Frampton ER, Schiff SD, et al. Genetic analysis of four consanguineous multiplex families with inflammatory bowel disease. Gastroenterology. 2021;9(6):521–532. doi:10.1093/gastro/goab007
12. Morsy Y, Brillant N, Franc Y, Scharl M, Wawrzyniak M; Swiss IBD Cohort Study Group. Unravelling the impact of the genetic variant rs1042058 within the TPL2 risk gene locus on molecular and clinical disease course patients with inflammatory bowel disease. Cells. 2021;10(12):3589. doi:10.3390/cells10123589
13. Zhang M, Wang X, Jiang X, et al. Polymorphisms of the TNF gene and three susceptibility loci are associated with Crohn’s disease and perianal Fistula Crohn’s disease: a study among the Han Population from South China. Med Sci Monit. 2019;25:9637. doi:10.12659/MSM.917244
14. Cleynen I, Boucher G, Luke Jostins L, et al. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. lancet. 2016;387(10014):156–167. doi:10.1016/S0140-6736(15)00465-1
15. Sidiq T, Yoshihama S, Downs I, Kobayashi KS. Nod2: a critical regulator of ileal microbiota and Crohn’s disease. Front Immunol. 2016;7:367. doi:10.3389/fimmu.2016.00367
16. Hampe J, Cuthbert A, Croucher PJP, et al. Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. lancet. 2001;357(9272):1925–1928. doi:10.1016/S0140-6736(00)05063-7
17. Nagy Z, Karádi O, Rumi G. Crohn’s disease is associated with polymorphism of CARD15/NOD2 gene in a Hungarian population. Ann N Y Acad Sci. 2005;1051(1):45–51. doi:10.1196/annals.1361.045
18. Cavanaugh JA, Adams KE, Quak EJ, et al. CARD15/NOD2 risk alleles in the development of Crohn’s disease in the Australian population. Ann Hum Genet. 2003;67(1):35–41. doi:10.1046/j.1469-1809.2003.00006.x
19. Arnott IDR, Nimmo ER, Drummond HE, et al. NOD2/CARD15, TLR4 and CD14 mutations in Scottish and Irish Crohn’s disease patients: evidence for genetic heterogeneity within Europe?. Genes Immun. 2004;5(5):417–425. doi:10.1038/sj.gene.6364111
20. Karban A, Atia O, Leitersdorf E, et al. The relation between NOD2/CARD15 mutations and the prevalence and phenotypic heterogeneity of Crohn’s disease: lessons from the Israeli Arab Crohn’s disease cohort. Dig Dis Sci. 2005;50(9):1692–1697. doi:10.1007/s10620-005-2917-x
21. Tolentino YFM, Elia PP, Fogaça HS, et al. Common NOD2/CARD15 and TLR4 polymorphisms are associated with Crohn’s disease phenotypes in southeastern Brazilians. Dig Dis Sci. 2016;61(9):2636–2647. doi:10.1007/s10620-016-4172-8
22. Leong RWL, Armuzzi A, Ahmad T, et al. NOD2/CARD15 gene polymorphisms and Crohn’s disease in the Chinese population. Aliment Pharmacol Ther. 2003;17(12):1465–1470. doi:10.1046/j.1365-2036.2003.01607
23. Ng SC, Tsoi KKF, Kamm MA, et al. Genetics of inflammatory bowel disease in Asia: systematic review and meta-analysis. Inflamm Bowel Dis. 2012;18(6):1164–1176. doi:10.1002/ibd.21845
24. Iqbal Siddique AS, Mustafa IK, Ziyab AH, et al. Detection of mutations in NOD2/CARD15 gene in Arab patients with Crohn’s disease. Saudi J Gastroenterol. 2021;27(4):240. doi:10.4103/sjg.sjg_582_20
25. Abdelnaby H, Ndiaye N, D’Amico F, et al. NOD2/CARD15 polymorphisms (P268S, IVS8+ 158, G908R, L1007fs, R702W) among Kuwaiti patients with Crohn’s disease: a case-control study. Saudi J Gastroenterol. 2021;27(4):249. doi:10.4103/sjg.sjg_613_20
26. Abu-Freha N, Badarna W, Sigal-Batikoff I, et al. ASCA and ANCA among Bedouin Arabs with inflammatory bowel disease, the frequency and phenotype correlation. BMC Gastroenterol. 2018;18(1):1–5. doi:10.1186/s12876-018-0884-x
27. Feki S, Bouzid D, Abida O, et al. Genetic association and phenotypic correlation of TLR4 but not NOD2 variants with Tunisian inflammatory bowel disease. J Dig Dis. 2017;18(11):625–633. doi:10.1111/1751-2980.12552
28. Giudici F, Cavalli T, Luceri C, et al. Long-term follow-up, association between CARD15/NOD2 polymorphisms, and clinical disease behavior in Crohn’s disease surgical patients. Mediators Inflamm. 2021;2021:1–11. doi:10.1155/2021/8854916
29. Kaczmarek-Ryś M, TytusHryhorowicz S, Lis E, et al. Crohn’s disease susceptibility and onset are strongly related to three NOD2 gene haplotypes. J Clin Med. 2021;10(17):3777. doi:10.3390/jcm10173777
30. Horowitz JE, Warner N, Staples J, et al. Mutation spectrum of NOD2 reveals recessive inheritance as a main driver of Early Onset Crohn’s Disease. Sci Rep. 2021;11(1):1–10. doi:10.1038/s41598-021-84938-8
31. Chuang AY, Chuang JC, Zhai Z, Feng W, Kwon JH. NOD2 expression is regulated by microRNAs in colonic epithelial HCT116 cells. Inflamm Bowel Dis. 2014;20(1):126–135. doi:10.1097/01.MIB.0000436954.70596.9b
32. Fachal L. The International IBD Genetics Consortium, on behalf of the International IBD Genetics Consortium, OP11 Expanded genome-wide association study of Inflammatory Bowel Disease identifies 174 novel loci and directly implicates new genes in disease susceptibility. J Crohns Colitis. 2022;16(1):i011–i013. doi:10.1093/ecco-jcc/jjab232.010
33. Turpin W, Goethel A, Bedrani L, Kenneth Croitoru MDCM. Determinants of IBD heritability: genes, bugs, and more. Inflamm Bowel Dis. 2018;24(6):1133–1148. doi:10.1093/ibd/izy085
34. Annese V. Genetics and epigenetics of IBD. Pharmacol Res. 2020;159:104892. doi:10.1016/j.phrs.2020.104892
35. Papoutsopoulou S, Campbell BJ. Epigenetic modifications of the nuclear factor kappa B signalling pathway and its impact on inflammatory bowel disease. Curr Pharm Des. 2021;27(35):3702–3713. doi:10.2174/1381612827666210218141847
36. Feuerstein JD, Cheifetz AS. Crohn disease: epidemiology, diagnosis, and management. Mayo Clinic Proceed. 2017;92(7):1088–1103. doi:10.1016/j.mayocp.2017.04.010
37. Robinson EL, Anene-Nzelu CG, Manuel R-G, Foo RSY. Cardiac epigenetics: driving signals to the cardiac epigenome in development and disease. J Mol Cell Cardiol. 2021;151:88. doi:10.1016/j.yjmcc.2020.11.005
38. Larabi A, Barnich N, Nguyen HTT. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy. 2020;16(1):38–51. doi:10.1080/15548627.2019.1635384
39. Kim S, Eun HS, Kyeong E. Roles of autophagy-related genes in the pathogenesis of inflammatory bowel disease. Cells. 2019;8(1):77. doi:10.3390/cells8010077
40. Takagawa T, Kitani A, Fuss I, et al. An increase in LRRK2 suppresses autophagy and enhances Dectin-1–induced immunity in a mouse model of colitis. Sci Transl Med. 2018;10(444):eaan8162. doi:10.1126/scitranslmed.aan8162
41. Netea-Maier RT, Plantinga TS, van de Veerdonk FL, Smit JW, Netea MG. Modulation of inflammation by autophagy: consequences for human disease. Autophagy. 2016;12(2):245–260. doi:10.1080/15548627.2015.1071759
42. de Bruyn M, Vermeire S. NOD2 and bacterial recognition as therapeutic targets for Crohn’s disease. Expert Opin Ther Targets. 2017;21(12):1123–1139. doi:10.1080/14728222.2017.1397627
43. Nambu R, Muise AM. Advanced understanding of monogenic inflammatory bowel disease. Front Pediatr. 2021;8:618918. doi:10.3389/fped.2020.618918
44. Uhlig HH, Schwerd T, Koletzko S, et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology. 2014;147(5):990–1007. doi:10.1053/j.gastro.2014.07.023
45. Skakic A, Djordjevic M, Sarajlija A, et al. Genetic characterization of GSD I in Serbian population revealed unexpectedly high incidence of GSD Ib and 3 novel SLC37A4 variants. Clin Genet. 2018;93(2):350–355. doi:10.1111/cge.13093
46. Imai K, Morio T, Zhu Y, et al. Clinical course of patients with WASP gene mutations. Blood. 2004;103(2):456–464. doi:10.1182/blood-2003-05-1480
47. Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet. 2013;132(10):1077–1130. doi:10.1007/s00439-013-1331-2
48. Liu JZ, Van Sommeren S, Hailiang Huang SC, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47(9):979–986. doi:10.1038/ng.3359
49. De Lange KM, Loukas Moutsianas JC, Lee CA, et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet. 2017;49(2):256–261. doi:10.1038/ng.3760
50. Shaw KA, Cutler DJ, Okou D, et al. Genetic variants and pathways implicated in a pediatric inflammatory bowel disease cohort. Genes Immun. 2019;20(2):131–142. doi:10.1038/s41435-018-0015-2
51. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14(5):329–342. doi:10.1038/nri3661
52. Miner-Williams WM, Moughan PJ. Intestinal barrier dysfunction: implications for chronic inflammatory conditions of the bowel. Nutr Res Rev. 2016;29(1):40–59. doi:10.1017/S0954422416000019
53. Chen S-J, Liu X-W, Liu J-P, Yang X-Y, Fang-Gen L. Ulcerative colitis as a polymicrobial infection characterized by sustained broken mucus barrier. World J Gastroenterol. 2014;20(28):9468. doi:10.3748/wjg.v20.i28.9468
54. Guan Q. A comprehensive review and update on the pathogenesis of inflammatory bowel disease. J Immunol Res. 2019;2019:1–16. doi:10.1155/2019/7247238
55. Chang JT. Pathophysiology of inflammatory bowel diseases. N Eng J Med. 2020;383(27):2652–2664. doi:10.1056/NEJMra2002697
56. Marlies M, Mayassi T, Fehlner-Peach H, et al. Interleukin-15 promotes intestinal dysbiosis with butyrate deficiency associated with increased susceptibility to colitis. ISME J. 2017;11(1):15–30. doi:10.1038/ismej.2016.114
57. Hodzic Z, Schill EM, Bolock AM, Good M. IL-33 and the intestine: the good, the bad, and the inflammatory. Cytokine. 2017;100:1–10. doi:10.1016/j.cyto.2017.06.017
58. Griesenauer B, Paczesny S. The ST2/IL-33 axis in immune cells during inflammatory diseases. Front Immunol. 2017;8:475. doi:10.3389/fimmu.2017.00475
59. Ni J, Wu GD, Albenberg L, Tomov VT. Gut microbiota and IBD: causation or correlation? Nature Rev. 2017;14(10):573–584. doi:10.1038/nrgastro.2017.88
60. Ahluwalia B, Moraes L, Magnusson MK, Öhman L. Immunopathogenesis of inflammatory bowel disease and mechanisms of biological therapies. Scand J Gastroenterol. 2018;53(4):379–389. doi:10.1080/00365521.2018.1447597
61. Wallace KL, Zheng L-B, Kanazawa Y, Shih DQ. Immunopathology of inflammatory bowel disease. World J Gastroenterol. 2014;20(1):6. doi:10.3748/wjg.v20.i1.6
62. Baumgart DC, Sandborn WJ. Crohn’s disease. lancet. 2012;380(9853):1590–1605. doi:10.1016/S0140-6736(12)60026-9
63. Manichanh C, Borruel N, Casellas F, Guarner F. The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol. 2012;9(10):599–608. doi:10.1038/nrgastro.2012.152
64. Zundler S, Neurath MF. Immunopathogenesis of inflammatory bowel diseases: functional role of T cells and T cell homing. Clin Exp Rheumatol. 2015;33(4):S19–S28.
65. De Souza HSP, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol. 2016;13(1):13–27. doi:10.1038/nrgastro.2015.186
66. Lee YK, Mazmanian SK. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science. 2010;330(6012):1768–1773. doi:10.1126/science.1195568
67. de Zoete MR, Flavell RA. Interactions between nod-like receptors and intestinal bacteria. Front Immunol. 2013;4:462. doi:10.3389/fimmu.2013.00462
68. Huldani H, Margiana R, Ahmad F, et al. Immunotherapy of inflammatory bowel disease (IBD) through mesenchymal stem cells. Int Immunopharmacol. 2022;107:108698. doi:10.1016/j.intimp.2022.108698
69. Petersen C, Round JL. Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol. 2014;16(7):1024–1033. doi:10.1111/cmi.12308
70. Cohen LJ, Cho JH, Gevers D, Chu H. Genetic factors and the intestinal microbiome guide development of microbe-based therapies for inflammatory bowel diseases. Gastroenterology. 2019;156(8):2174–2189. doi:10.1053/j.gastro.2019.03.017
71. Mishima Y, Sartor RB. Manipulating resident microbiota to enhance regulatory immune function to treat inflammatory bowel diseases. J Gastroenterol. 2020;55(1):4–14. doi:10.1007/s00535-019-01618-1
72. Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474(7351):307–317. doi:10.1038/nature10209
73. López L, Rosario MJ, Burgos G, Gálvez A, Pulido RP. The human gastrointestinal tract and oral microbiota in inflammatory bowel disease: a state of the science review. Apmis. 2017;125(1):3–10. doi:10.1111/apm.12609
74. Halfvarson J, Brislawn CJ, Lamendella R, et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nature Microbiol. 2017;2(5):1–7. doi:10.1038/nmicrobiol.2017.4
75. Santana PT, Rosas SLB, Ribeiro BE, Marinho Y, de Souza HSP. Dysbiosis in inflammatory bowel disease: pathogenic role and potential therapeutic targets. Int J Mol Sci. 2022;23(7):3464. doi:10.3390/ijms23073464
76. Mahadevan U. Fertility and pregnancy in the patient with inflammatory bowel disease. Gut. 2006;55(8):1198–1206. doi:10.1136/gut.2005.078097
77. Kennedy KM, Gerlach MJ, Adam T, et al. Fetal meconium does not have a detectable microbiota before birth. Nature Microbiol. 2021;6(7):865–873. doi:10.1038/s41564-021-00904-0
78. Koren O, Goodrich JK, Cullender TC, et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell. 2012;150(3):470–480. doi:10.1016/j.cell.2012.07.008
79. Wang J, Shi Z-H, Yang J, Wei Y, Wang X-Y, Zhao Y-Y. Gut microbiota dysbiosis in preeclampsia patients in the second and third trimesters. Chin Med J. 2020;133(9):1057–1065. doi:10.1097/CM9.0000000000000734
80. Nuriel-Ohayon M, Neuman H, Ziv O, et al. Progesterone increases Bifidobacterium relative abundance during late pregnancy. Cell Rep. 2019;27(3):730–736. doi:10.1016/j.celrep.2019.03.075
81. Simone D, Nicoletta A, Ortiz S, et al. Recent insights on the maternal microbiota: impact on pregnancy outcomes. Front Immunol. 2020;2372. doi:10.3389/fimmu.2020.528202
82. Torres J, Hu J, Seki A, et al. Infants born to mothers with IBD present with altered gut microbiome that transfers abnormalities of the adaptive immune system to germ-free mice. Gut. 2020;69(1):42–51. doi:10.1136/gutjnl-2018-317855
83. de Agüero G, Ganal-Vonarburg M, Fuhrer SC, et al. The maternal microbiota drives early postnatal innate immune development. Science. 2016;351(6279):1296–1302. doi:10.1126/science.aad2571
84. Dominguez-Bello MG, De Jesus-Laboy KM, Shen N, et al. Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer. Nat Med. 2016;22(3):250–253. doi:10.1038/nm.4039
85. Peter I, Maldonado-Contreras A, Eisele C, et al. A dietary intervention to improve the microbiome composition of pregnant women with Crohn’s disease and their offspring: the MELODY (Modulating Early Life Microbiome through Dietary Intervention in Pregnancy) trial design. Contemp Clin Trials Commun. 2020;18:100573. doi:10.1016/j.conctc.2020.100573
86. Gueimonde M, Sakata S, Kalliomäki M, Isolauri E, Benno Y, Salminen S. Effect of maternal consumption of lactobacillus GG on transfer and establishment of fecal bifidobacterial microbiota in neonates. J Pediatr Gastroenterol Nutr. 2006;42(2):166–170. doi:10.1097/01.mpg.0000189346.25172.fd
87. Stinson LF, Boyce MC, Payne MS, Keelan JA. The not-so-sterile womb: evidence that the human fetus is exposed to bacteria prior to birth. Front Microbiol. 2019;10:1124. doi:10.3389/fmicb.2019.01124
88. Gilbert SF, Bosch TC, Ledón-Rettig C. Eco-Evo-Devo: developmental symbiosis and developmental plasticity as evolutionary agents. Nature Rev. 2015;16(10):611–622. doi:10.1038/nrg3982
89. He Q, Kwok L-Y, Xiaoxia X, et al. The meconium microbiota shares more features with the amniotic fluid microbiota than the maternal fecal and vaginal microbiota. Gut Microbes. 2020;12(1):1794266. doi:10.1080/19490976.2020.1794266
90. Galazzo G, van Best N, Bervoets L, et al. Development of the microbiota and associations with birth mode, diet, and atopic disorders in a longitudinal analysis of stool samples, collected from infancy through early childhood. Gastroenterology. 2020;158(6):1584–1596. doi:10.1053/j.gastro.2020.01.024
91. Meyer A, Drouin J, Weill A, Carbonnel F, Dray-Spira R. Pregnancy in women with inflammatory bowel disease: a French nationwide study 2010–2018. Aliment Pharmacol Ther. 2020;52(9):1480–1490. doi:10.1111/apt.16074
92. Hill JE, Peña-Sánchez J-N, Champika Fernando AC, et al. Composition and stability of the vaginal microbiota of pregnant women with inflammatory bowel disease. Inflamm Bowel Dis. 2021. doi:10.1093/ibd/izab314
93. Cai Z, Wang S, Jiannan L. Treatment of inflammatory bowel disease: a comprehensive review. Front Med. 2021;2681. doi:10.3389/fmed.2021.765474
94. Sveiven SN, Nordgren TM. Lung-resident mesenchymal stromal cells are tissue-specific regulators of lung homeostasis. Am J Physiol Lung Cell Mol. 2020;319(2):L197–L210. doi:10.1152/ajplung.00049.2020
95. Spiewak TA, Patel A. User’s guide to JAK inhibitors in inflammatory bowel disease. Current Res Pharmacol Drug Discov. 2022;3:100096. doi:10.1016/j.crphar.2022.100096
96. Gottlieb ZS, Sands BE. Personalized medicine with IL-23 blockers: myth or reality? J Crohns Colitis. 2021. doi:10.1093/ecco-jcc/jjab190
97. Scarozza P, Schmitt H, Monteleone G, Neurath MF, Atreya R. Oligonucleotides—a novel promising therapeutic option for IBD. Front Pharmacol. 2019;10:314. doi:10.3389/fphar.2019.00314
98. Hryhorowicz S, Kaczmarek-Ryś M, Aleksandra Zielińska RJ, Scott RS, Andrzej P. Endocannabinoid system as a promising therapeutic target in inflammatory bowel disease–a systematic review. Front Immunol. 2021;12:790803. doi:10.3389/fimmu.2021.790803
99. Li Y-H, Sun W, Zhou B-J, et al. iTRAQ-based pharmacoproteomics reveals potential targets of berberine, a promising therapy for ulcerative colitis. Eur J Pharmacol. 2019;850:167–179. doi:10.1016/j.ejphar.2019.02.021
100. Delmondes LM, Nunes MO, Azevedo AR, Oliveira MM, Coelho LE, Torres-Neto JD. Clinical and sociodemographic aspects of inflammatory bowel disease patients. Gastroenterol Res. 2015;8(3–4):207–215. doi:10.14740/gr649w
101. Spekhorst LM, Imhann F, Festen EAM, et al. Cohort profile: design and first results of the Dutch IBD Biobank: a prospective, nationwide biobank of patients with inflammatory bowel disease.. BMJ open. 2017;7(11):e016695. doi:10.1136/bmjopen-2017-016695
102. Wang P, Jun H, Xiao A, et al. Smoking and inflammatory bowel disease: a comparison of China, India, and the USA. Dig Dis Sci. 2018;63(10):2703–2713. doi:10.1007/s10620-018-5142-0
103. Bastida G, Beltrán B. Ulcerative colitis in smokers, non-smokers and ex-smokers. World J Gastroenterol. 2011;17(22):2740–2747. doi:10.3748/wjg.v17.i22.2740
104. Nos P, Domènech E. Management of Crohn’s disease in smokers: is an alternative approach necessary?. World J Gastroenterol. 2011;17(31):3567. doi:10.3748/wjg.v17.i31.3567
105. AlQasrawi D, Abdelli LS, Naser SA. mystery solved: why smoke extract worsens disease in smokers with Crohn’s Disease and Not Ulcerative Colitis? Gut MAP!. Microorganisms. 2020;8(5):666. doi:10.3390/microorganisms8050666
106. Mohammed HO, Ahmed Alaa El-Din E, Farag AI. Impact of e-cigarettes on colonic mucosa and the role of recovery: involvement of oxidative and inflammatory pathway. Environ Sci Pollut Res Int. 2021;28(45):64561–64571. doi:10.1007/s11356-021-15575-x
107. Olendzki BC, Silverstein TD, Persuitte GM, Yunsheng M, Baldwin KR, Cave D. An anti-inflammatory diet as treatment for inflammatory bowel disease: a case series report. Nutr J. 2014;13(1):1–7. doi:10.1186/1475-2891-13-5
108. Coelho MR, DiogoRomi M, Ferreira DM, Zaltman C, Soares-Mota M. The use of curcumin as a complementary therapy in ulcerative colitis: a systematic review of randomized controlled clinical trials. Nutrients. 2020;12(8):2296. doi:10.3390/nu12082296
109. Yang Z, Liu W, Zhou X, Zhu X, Suo F, Yao S. The effectiveness and safety of curcumin as a complementary therapy in inflammatory bowel disease: a protocol of systematic review and meta-analysis. Medicine. 2020;99(43). doi:10.1097/MD.0000000000022916
110. Rizzello F, Spisni E, Giovanardi E, et al. Implications of the westernized diet in the onset and progression of IBD. Nutrients. 2019;11(5):1033. doi:10.3390/nu11051033
111. Bischoff NS, de Kok TM, Sijm DTHM, et al. Possible adverse effects of food additive E171 (titanium dioxide) related to particle specific human toxicity, including the immune system. Int J Mol Sci. 2020;22(1):207. doi:10.3390/ijms22010207
112. Khalili H, Ananthakrishnan AN, Konijeti GG, et al. Physical activity and risk of inflammatory bowel disease: prospective study from the Nurses’ Health Study cohorts. BMJ. 2013;347. doi:10.1136/bmj.f6633
113. Dong J, Chen Y, Tang Y, et al. Body mass index is associated with inflammatory bowel disease: a systematic review and meta-analysis. PLoS One. 2015;10(12):e0144872. doi:10.1371/journal.pone.0144872
114. Fletcher J, Cooper SC, Ghosh S, Hewison M. The role of vitamin D in inflammatory bowel disease: mechanism to management. Nutrients. 2019;11(5):1019. doi:10.3390/nu11051019
115. Bosman ES, Albert AY, Harvey Lui JP. Skin exposure to narrow band ultraviolet (UVB) light modulates the human intestinal microbiome. Front Microbiol. 2019;10:2410. doi:10.3389/fmicb.2019.02410
116. Teich N, Bruns T, Stallmach A. Appendectomy in childhood—did it save my sibling from getting ulcerative colitis? Int J Colorectal Dis. 2021;36(3):623–624. doi:10.1007/s00384-020-03780-y
117. Chung W-S, Chung S, Hsu C-Y, Lin C-L. Risk of inflammatory bowel disease following appendectomy in adulthood. Front Med. 2021;8:808. doi:10.3389/fmed.2021.661752
118. Gubatan J, Levitte S, Patel A, et al. Artificial intelligence applications in inflammatory bowel disease: emerging technologies and future directions. World J Gastroenterol. 2021;27(17):1920. doi:10.3748/wjg.v27.i17.1920
119. Chatterji M, Fidler JL, Taylor SA, Anupindi SA, Yeh BM, Guglielmo FF. State of the art MR enterography technique. Topics Magn Reson Imaging. 2021;30(1):3–11. doi:10.1097/RMR.0000000000000263
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.