")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 17
A Sporadic Family of Lipoid Proteinosis with Novel ECM1 Gene Mutations
Authors Liu YL, Zhang ZYO, Chen XM
Received 19 December 2023
Accepted for publication 31 March 2024
Published 18 April 2024 Volume 2024:17 Pages 885—889
DOI https://doi.org/10.2147/CCID.S452127
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Rungsima Wanitphakdeedecha
Yu-Ling Liu, Zeng-Yun-Ou Zhang, Xiao-Mei Chen
Department of Dermatology and Venerology, West China Hospital of Sichuan University, Chengdu, People’s Republic of China
Correspondence: Xiao-Mei Chen, Department of Dermatology, West China Hospital of Sichuan University, Chengdu, People’s Republic of China, Email [email protected]
Abstract: Lipoid proteinosis (LP) is an uncommon, autosomal recessive genetic disorder. Multigene panel testing was conducted to confirm the diagnosis of a sporadic family with suspected LP. In the proband, we identified two mutations of ECMI and provided genetic evidence for informed genetic counselling.
Keywords: proteinosis, lipoid, multigene panel testing, ECM1 gene
Introduction
Lipoid proteinosis (LP), also known as Urbach-Wiethe syndrome, is a rare autosomal recessive genetic disorder characterized by the presence of superficial beaded pearls along the eyelids and waxy papules on the skin.1 Histological examination reveals abnormal deposition of proteins and lipids in the dermis and deep dermis. LP is caused by a homozygous or compound heterozygous loss-of-function mutation of extracellular matrix protein 1 (ECM1), which is located on chromosome 1q21.2 To date, more than 40 different variants of ECM1 mutations have been reported, most of which are nonsense or missense mutations located in exons 6 and 7. In this study, we report a case of LP caused by novel ECM1 gene mutations.
Case Report
A 5-year-old Chinese boy, presented with beaded papules on the eyelids and atrophic scars throughout the body upon admission to our institution. Normal skin was observed at birth; however, atrophic scars developed on the neck, shoulders, and buttocks due to easy bruising and festering. Hoarseness of voice began at 2 years old followed by gradual onset of dysphagia and obstruction. Hyperplastic gums and tongues were noted by age 3. Verrucous hyperplastic lesions on the fingertips when he turned 4. The patient did not receive regular treatment for his condition. No abnormalities in vision, epilepsy, psychosis or dyskinesia were found in the patient, nor was there any significant difference in intelligence compared to his peers. The parents were non-consanguineous without any family history of similar conditions.
During physical examination, beaded papules were observed along the upper eyelid margin (Figure 1A), accompanied by disordered arrangement of teeth and gingival hyperplasia (Figure 1B). Diffuse atrophic scars covered his entire body (Figure 1C), while brownish-yellow plaques were present on both knees (Figure 1D). Dermoscopy revealed clustered light yellow oval structureless areas on the upper eyelids (Figure 1E), as well as verrucous keratinized proliferative lesions around the nails (Figure 1F). Laryngoscopic examination showed a granulomatous mass located posteriorly to the left vocal cord along with partial defect of the right vocal cord and weakened mucosal waves bilaterally.
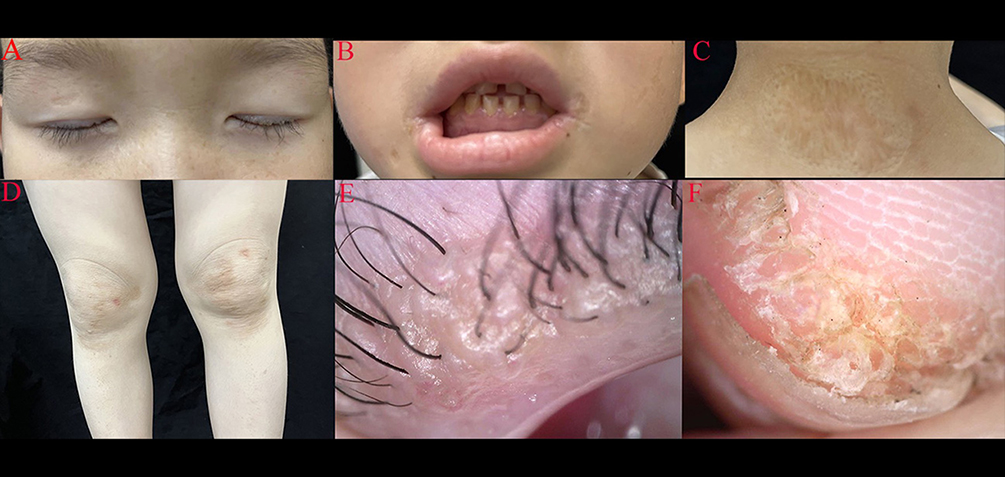 |
Figure 1 Physical examination revealed beaded papules on the upper eyelid margin (A), disordered arrangement of teeth and gingival hyperplasia (B), diffused atrophic scar on the whole body (C), and brownish-yellow plaques on both knees (D), clustered light yellow oval structureless area on the upper eyelids (dermoscopy) (E), verrucous, keratinized, proliferative lesions around the nails (F). |
After obtaining informed consent from the parents, peripheral blood samples were collected from the patient and both parents for multigene panel testing. DNA was extracted using the Qiagen Blood DNA mini kit (Qiagen, Germany). The extracted DNA underwent multigene panel testing on the DNBSEQ-T7 sequencer (MGI, Shenzhen, China), with a focus on analyzing 707 genes associated with hereditary dermatoses.
Low-quality sequences were filtered using CutAdapter software (https://cutadapt.readthedocs.io/en/stable/). The remaining reads were aligned to the human genome version hg19 using the BWA aligner (https://github.com/lh3/bwa). The annotation of SNPs and INDELs was performed using ANNOVAR (https://www.openbioinformatics.org/annovar/), with a focus on mutation sites present at frequencies less than 0.02 in the normal database, which include data from the Thousand Genomes Project (https://www.internationalgenome.org/), Exome Variant Server (https://evs.gs.washington.edu/EVS/), and Exome Aggregation Consortium (EXAC) (https://www.broadinstitute.org/exac).
Subsequently, the genomic DNA isolated from the proband and his relatives underwent Sanger sequencing. This sequencing, in conjunction with the multigene panel testing, unveiled two previously unreported heterozygous variants in the ECM1 gene within this family. In both the proband and his father (Figure 2A and B), a novel frameshift variant designated as c.317_339delTCCCTCAGGAAGCTGTCCCCCTC was identified in exon 5, resulting in a Leu106Profs*12 amino acid change that introduced a frameshift and produced an aberrant ECM1 protein. Furthermore, in the proband and his mother (Figure 2C and D), a nonsense variant designated as c.479G>A was detected in exon 6 leading to a Trp160X amino acid change that prematurely truncated the protein by introducing a stop codon at position 160. Importantly, neither of these variants were present in DNA samples collected from 100 unaffected healthy controls. LP was diagnosed based on characteristic clinical findings and multigene panel testing.
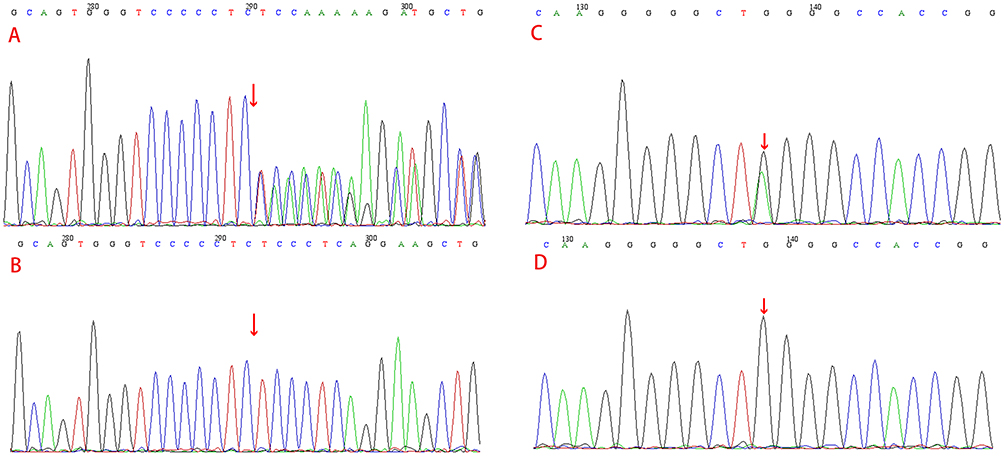 |
Figure 2 Mutational analysis of this family. The red arrows indicate the mutation sites. The c.317_339delTCCCTCAGGAAGCTGTCCCCCTC:p.Leu106Profs*12 frameshift variant in ECM1 in exon 5 of the proband and his father (A); there was no mutation in his mother (B); c.479G>A:p. Trp160X nonsense variant in ECM1 in exon 6 of the proband and his mother (C); no mutation was detected in his father (D). |
Discussion
LP was first discovered and described by Urbach and Wiethe in 1929.1,2 This disorder is characterized by abnormal deposition of lipids and proteins in the dermis and submucosal connective tissue. To date, fewer than 500 cases have been documented in the literature.3–5 Homozygous or compound heterozygous mutations in the ECM1 gene located on chromosome 1q21 are believed to be responsible for this condition.3 The ECM1 gene encodes a glycoprotein that functions as a transporter and binds to growth and differentiation factors. Mutations in ECM1 leading to aberrant protein expression result in the deposition of hyaline material in various organs, including the mucous membrane and skin, potentially contributing to the diverse clinical manifestations observed.
Hoarseness, caused by infiltration of transparent substances into the vocal cords, is typically the initial clinical manifestation in LP.4–6 Additionally, ocular, laryngeal, and neurological involvement may also occur.7 The cutaneous manifestations of LP are diverse and often manifest within the first two years of life and present as two overlapping stages. The first stage involves the development of vesicular, bullous, or erosive lesions due to minor trauma or friction, which heal with scarring on the face, dorsum of the hands, and forearm. The second stage is characterized by the gradual emergence of verrucous lesions, on the extensor surfaces of the elbows, hands, and knees, as well as diffuse skin thickening, hyperkeratosis and nodules.8 Classical skin signs of LP consist of multiple beaded papules along the eyelid margins and inner canthus.9 Furthermore, various systemic manifestations have been described such as seizures, psychiatric disorders, memory loss, gingival hyperplasia, stiff tongue and slurred speech.3,10,11 There have been reports of disabling tongue ulcers developing among LP patients, which improve following oral corticosteroid treatment, resulting in gradual epithelization of the ulcer.12
LP is frequently observed in offspring of consanguineous marriages. A study revealed that 20% of LP probands’ parents were from consanguineous unions.13 Multigene Panel targeted testing greatly enhances the diagnostic accuracy for genetic diseases like LP, pinpointing pathogenic mutations through precise gene sequencing of the proband and their parents. This approach minimizes physical trauma and scarring risks associated with biopsies, ensuring reliable results even with young and uncooperative patients. ECM1, identified as a pathogenic gene for LP in 2002,14 has now been associated with over 40 different mutations, predominantly nonsense or missense mutations located in exons 6 and 7. In our study, compound heterozygous mutations were detected in this family: one frameshift variant (c.317_339delTCCCTCAGGAAGCTGTCCCCCTC: p.Leu106Profs*12) in exon 5 and one nonsense variant (c.479G>A: p.Trp160X) in exon 6 of ECM1, potentially leading to abnormal protein expression. The proband carried two mutant ECM1 genes while his parents had heterozygous variants. These novel mutations were classified as PSV1 with strong evidence supporting their pathogenicity grade. These findings align well with the clinical phenotypes exhibited by the proband.
However, the genotype-phenotype correlations remain unclear due to potential differential activity of several ECM1 transcripts across various tissues.4,15 In clinics, LP patients vary in clinical manifestations, showing different degrees of skin, mucosal scarring and infiltration.15,16 We aim to clarify the link between gene mutations and these phenotypes to better understand LP’s nature and find more effective treatments in future. Long-term follow-up will also help identify and manage complications, improving patients’ quality of life and reducing psychological issues.
Although there is currently no cure for LP, it does not impact the normal life expectancy of patients.17 Various treatment modalities have been employed for managing skin lesions, including carbon dioxide lasers, etretinate, acitretin, penicillamine, dimethyl sulfoxide, surgical procedures and dermabrasion.7,18,19 Nearly all patients experience laryngeal involvement, and tracheotomy should be considered in cases where severe airway problems are present.6 In this particular case study, the surgeon recommended surgical intervention at the age of 7 years to address vocal cord issues. Due to financial constraints, no specific treatment was administered for the skin lesions. The patient continues to receive regular follow-up care.
In summary, we confirmed the diagnosis of LP in a sporadic family by conducting multigene panel testing. These novel pathogenic variants expand the spectrum of ECM1 pathogenic variants and provide genetic evidence for genetic counseling.
The ethical committee of the hospital gave the agreement to report this case.
Consent Statement
A formal written consent was obtained from the patient’s parents for publication the case details and associated images from the patient.
Acknowledgments
We thank the patient and his family for their participation in the study.
Disclosure
The authors have no conflicts of interest to declare for this work.
References
1. Wei Z, Labbe A, Liang Q. Lipoid Proteinosis presenting as beaded papules of the eyelid: report of three cases. BMC Ophthalmol. 2021;21(1):35. doi:10.1186/s12886-021-01802-z
2. Hamada T, McLean WI, Ramsay M, et al. Lipoid proteinosis maps to 1q21 and is caused by mutations in the extracellular matrix protein 1 gene (ECM1). Hum Mol Genet. 2002;11(7):833–840. doi:10.1093/hmg/11.7.833
3. McGrath JA. Lipoid proteinosis. Handb Clin Neurol. 2015;132:317–322.
4. Chan I, Liu L, Hamada T, et al. The molecular basis of lipoid proteinosis: mutations in extracellular matrix protein 1. Exp Dermatol. 2007;16(11):881–890. doi:10.1111/j.1600-0625.2007.00608.x
5. Bhattacharjee R, Chatterjee D, Vinay K. Lipoid Proteinosis. JAMA Dermatol. 2018;154(12):1479–1480. doi:10.1001/jamadermatol.2018.3435
6. Loos E, Kerkhofs L, Laureyns G. Lipoid proteinosis: a rare cause of hoarseness. J Voice. 2019;33(2):155–158. doi:10.1016/j.jvoice.2017.05.024
7. Shah JS, Shah HA. Lipoid proteinosis: review of Indian cases. J Oral Maxillofac Pathol. 2022;26(2):236–241. doi:10.4103/jomfp.jomfp_249_21
8. Sabater-Abad J, Matellanes-Palacios M, Pont-Sanjuan V, Miquel-Miquel J, Navarro-Conde P, Gimeno-Carpio E. Oral ulcer-a disabling manifestation in a patient with lipoid proteinosis. JAMA Dermatol. 2019;155(8):977–979. doi:10.1001/jamadermatol.2019.0805
9. Sellami D, Masmoudi A, Turki H, et al. Manifestations ophtalmologiques de la hyalinose cutanéo-muqueuse [Ophthalmic manifestations of lipoid proteinosis]. Presse Med. 2006;35(5 Pt 1):796–798. French. doi:10.1016/S0755-4982(06)74693-0
10. Appenzeller S, Chaloult E, Velho P, et al. Amygdalae calcifications associated with disease duration in lipoid proteinosis. J Neuroimaging. 2006;16(2):154–156. doi:10.1111/j.1552-6569.2006.00018.x
11. Teive HA, Pereira ER, Zavala JAA, et al. Generalized dystonia and striatal calcifications with lipoid proteinosis. Neurology. 2004;63(11):2168–2169. doi:10.1212/01.WNL.0000145602.64073.C2
12. Pilz AC, Zink A, Franz R, et al. Pyoderma gangrenosum in a patient with lipoid proteinosis (Urbach-Wiethe disease). J Eur Acad Dermatol Venereol. 2019;33(8):e293–e295. doi:10.1111/jdv.15564
13. Frenkel B, Vered M, Taicher S, et al. Lipoid proteinosis unveiled by oral mucosal lesions: a comprehensive analysis of 137 cases. Clin Oral Investig. 2017;21(7):2245–2251. doi:10.1007/s00784-016-2017-7
14. Hamada T. Lipoid proteinosis. Clin Exp Dermatol. 2002;27(8):624–629. doi:10.1046/j.1365-2230.2002.01143.x
15. Hamada T, Wessagowit V, South AP, et al. Extracellular matrix protein 1 gene (ECM1) mutations in lipoid proteinosis and genotype-phenotype correlation. J Invest Dermatol. 2003;120(3):345–350. doi:10.1046/j.1523-1747.2003.12073.x
16. Hofer PA. Urbach-Wiethe disease (lipoglycoproteinosis; lipoid proteinosis, hyalinosis cutis et mucosae). A clinico-genetic study of 14 families from northern Sweden. Hereditas. 1974;77(2):209–218. doi:10.1111/j.1601-5223.1974.tb00934.x
17. Hortensius R, Terburg D, Morgan B, et al. The dynamic consequences of amygdala damage on threat processing in Urbach-Wiethe Disease. A commentary on Pishnamazi et al. (2016). Cortex. 2017;88:192–197. doi:10.1016/j.cortex.2016.07.013
18. Vahidnezhad H, Youssefian L, Uitto J. Lipoid proteinosis. In: GeneReviews(®). Adam MP, Feldman J, Mirzaa GM, et al. editor, Seattle: University of Washington, Seattle Copyright © 1993–2024, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved; 1993.
19. Mainali S, Nayak R, Gaur S. Oral findings in a child with lipoid proteinosis: a case report and review. J Indian Soc Pedod Prev Dent. 2011;29(1):62–67. doi:10.4103/0970-4388.79946
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.