")
Back to Journals » Journal of Blood Medicine » Volume 14
Antithrombin Deficiency and Thrombosis: A Wide Clinical Scenario Reported in a Single Institution
Authors Marco-Rico A , Marco-Vera P
Received 20 April 2023
Accepted for publication 11 August 2023
Published 1 September 2023 Volume 2023:14 Pages 499—506
DOI https://doi.org/10.2147/JBM.S416355
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Ana Marco-Rico,1,2 Pascual Marco-Vera2,3
1Hemostasis and Thrombosis Department, Hematology Service, University General Hospital Dr. Balmis, Alicante, Spain; 2Biomedical Research Institute (ISABIAL), Alicante, Spain; 3Clinical Medicine Department, Miguel Hernández University, Alicante, Spain
Correspondence: Ana Marco-Rico, Hemostasis and Thrombosis Department, Hematology Service, University General Hospital Dr. Balmis, Pintor Baeza Avenue, 12, Alicante, 03010, Spain, Tel +34 965913863, Fax +34 965913869, Email [email protected]
Abstract: Congenital antithrombin (AT) deficiency represents the form of thrombophilia with the highest thrombotic risk. It is characterized by a heterogeneous clinical presentation, depending mostly on the family history of thrombosis and type of genetic mutation. Inherited AT deficiency promotes idiopathic thrombosis at an early age (even in the pediatric population) and at atypical sites. Therefore, a positive family background necessitates ruling out this high-risk thrombophilia at a young age. Studying first-degree relatives, even if they are asymptomatic, is essential to establish thromboprophylaxis and a proper therapeutic approach in case of thrombosis. Patients with congenital AT deficiency require indefinite anticoagulation owing to the high thrombotic recurrence rate. Here, we present four unrelated cases reported in our institution who were diagnosed with hereditary AT deficiency, with a contrasting clinical evolution.
Keywords: hereditary antithrombin deficiency, thrombosis, anticoagulation, heparin resistance, SERPINC1 gene mutation
Introduction
Antithrombin (AT) is a physiological natural anticoagulant that inhibits procoagulant serine proteases, such as activated factor II, IX, X and XI.1 In 1965, Egeberg reported the first family with AT deficiency and venous thrombosis (VT), with a decrease in both AT and heparin cofactor activity to approximately half the normal level.2 A low level of AT activity disrupts the hemostatic balance and, therefore, promotes thrombosis.3
Hereditary AT deficiency is an autosomal dominant condition, with an estimated prevalence in the general population between 0.02% and 0.2%. In patients who develop venous thromboembolic disease, either VT or pulmonary embolism (PE), the prevalence varies between 1% and 5%.4,5 Nearly 60% of the thrombotic events are unprovoked, while the remaining 40% are associated with a transient risk factor.6 Hereditary AT deficiency is associated with the highest risk of thrombosis.4,5
Quantitative (type I) and qualitative (type II) defects have been described. In type I, the level of AT antigen and its activity are similarly reduced,7 whereas in type II variants, the AT antigen level remains normal, but the AT activity is reduced as the result of a dysfunctional protein.8 Regarding the frequency of each type in people with inherited AT deficiency, although type II is much more frequent in the general population (12% with type I vs 88% with type II), type I is more prevalent in patients who develop thrombosis (60% with type I vs 40% with type II).9 This is the reason why type II could be underdiagnosed, especially if there is no family history of thrombosis (see Table 1).
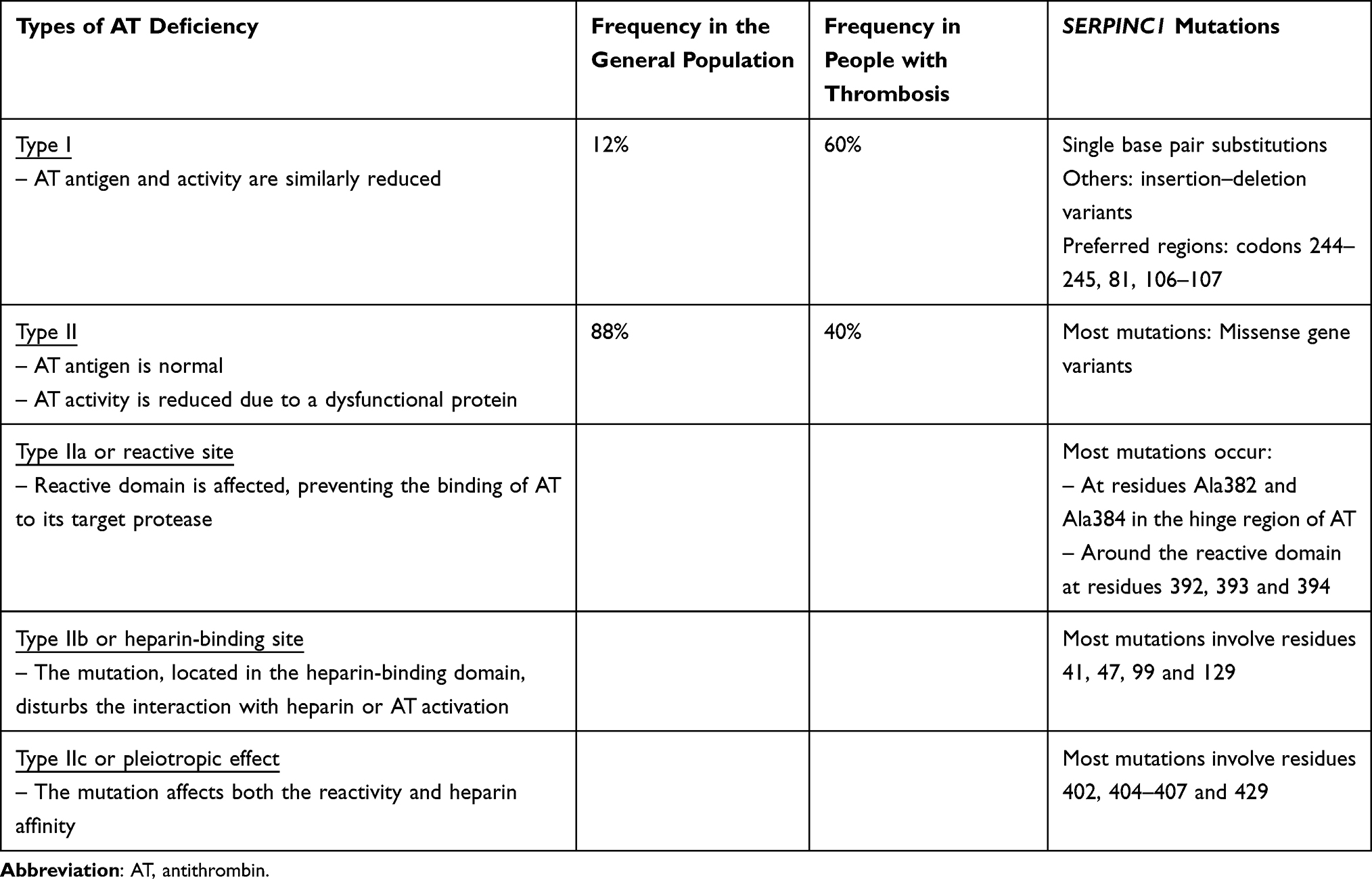 |
Table 1 Inherited AT Deficiency: Types, Frequency of Each Type in the General Population and in People with Thrombosis, and SERPINC1 Mutations |
The alteration of a single allele of SERPINC1 is the most frequent genetic mutation found in AT deficiency (352 genetic variants have been described).8 Most of the SERPINC1 genetic variants are point mutations (226 of 352 genetic variants). (See Table 1 for the most common mutations in the SERPINC1 gene, depending on the type of AT deficiency.) However, other mutations that affect the N-glycosylation pathway have been reported (up to 27%). These defects (hypoglycosylation) reduce the levels and the anticoagulant activity of AT as a result of an increase in 3‑N‑glycan AT in plasma.10
Clinical presentation may vary, depending mainly on the family history of thrombosis and type of genetic mutation. Type I AT-deficient patients usually develop thrombosis at an early age and have a tendency to recurrence. In contrast, type II AT-deficient patients have a wide clinical thrombotic presentation, ranging from mild to very severe. Of note, type IIb, or heparin-binding site mutation, carries a lower thrombotic risk.8
Homozygous AT deficiency type I is incompatible with life, while few cases of homozygous individuals with type IIb or mild type IIc have been described.8,9 Most patients with inherited heterozygous AT deficiency have an AT activity between 40% and 60%. Whether the risk of thrombosis correlates with the decrease in AT activity is still a matter of debate.9
The association with other additional genetic variants, such as factor V Leiden or the prothrombin G20210A mutation, increases the thrombotic risk, as does the presence of simultaneous acquired thrombotic risk factors, including immobilization, oral contraceptive intake, surgery, pregnancy, cancer and obesity.6
De la Morena-Barrio et al described an association between inferior vena cava atresia (IVCA) and congenital AT deficiency caused by the homozygous SERPINC1 c.391C>T variant (p.Leu131Phe; antithrombin Budapest 3). They included 61 patients with this mutation from a total of 1118 patients with congenital AT deficiency from Spain and Hungary. More than 70% of patients with available computed tomography (CT) had an atresia of the inferior vena cava (IVC) system. The authors suggested that thrombosis in the developing fetal vessels could be the reason for this anomaly.11
Additional case reports have described combined IVCA and congenital AT deficiency. According to these publications, ultrasound screening in newborns with relatives with IVCA and congenital AT deficiency is suggested, and early therapeutic intervention is necessary to prevent further thrombotic complications.12,13
Age is an important risk factor for thrombosis development. Up to 60% of patients with inherited AT deficiency have thrombosis before the age of 65.14 De la Morena-Barrio et al designed a multicenter study to determine the prevalence of thrombosis and presented clinical data in the pediatric population with inherited AT deficiency.14 The incidence of thrombosis in this population was 4.32 cases/1000 patients per year (7.5%), much higher than the incidence described in the general population (0.014%/year). They observed thrombosis at atypical sites such as the splanchnic vein, cerebral venous sinus or IVC, mainly in neonates and young children (<6 years old). Adolescents usually developed VT in lower limb and PE, and nearly half of them (47%) were provoked.14 An increased risk of first VT in AT-deficient subjects compared to controls (OR 16.26, 95% CI 9.90–26.70; P<0.00001) has been reported.15 An estimated annual risk of VT of up to 2.3% (95% CI 0.2–6.5) has been described.15 In addition, the annual risk of recurrence without long-term anticoagulant therapy was 8.8% (95% CI 4.6–14.1) for AT-deficient and 4.3% (95% CI 1.5–7.9) for non-AT-deficient VT patients (but with other types of thrombophilia).16
Patients with inherited AT deficiency who develop thrombosis require indefinite anticoagulation owing to the high thrombotic recurrence rate.16 Anticoagulation with vitamin K antagonists (VKAs) is the treatment of choice. AT deficiency induces heparin resistance. Therefore, reaching a therapeutic level of anti-Xa in patients anticoagulated with heparin can be challenging, despite the administration of the appropriate heparin dose adjusted for body weight. Using AT concentrates is a feasible option, together with heparin at therapeutic doses to treat acute thrombotic episodes and prevent new ones.5,17 Clinical evidence on the use of direct oral anticoagulants (DOACs) in AT-deficient patients is lacking. Case studies suggest that DOACs do not seem to be less effective than VKAs. No recurrences have been reported.18–20
Ten different families with hereditary AT deficiency are registered at our institution. Most of the family members require indefinite anticoagulation owing to thrombosis development.
Here, we present four unrelated patients with inherited AT deficiency who have been diagnosed and are being managed in our institution, as a reflection of the heterogeneous clinical presentation and individualized management.
Case 1 (C1)
Adult Woman with Venous Free-Floating Thrombus Despite Correct Anticoagulation
C1 is a 47-year-old woman diagnosed with AT deficiency type I (AT activity of 45% and AT antigen of 40%, confirmed in two different samples) in her childhood due to the known genetic background (father, aunts and cousins with AT deficiency and thrombosis). In all cases described in this manuscript, AT antigen was determined by immunoturbidimetry (Liatest ATIII®; Stago, Paris, France), while AT activity was measured using a factor II (bovine thrombin)-based chromogenic method (STA®-STACHROM® ATIII; Stago, Paris, France).
She had required indefinite anticoagulation since she developed a proximal deep VT in the left leg, confirmed by compression ultrasound, in the 15th week of pregnancy despite thromboprophylaxis with low-molecular-weight heparin (LMWH). She had no cardiovascular risk factors (CVRFs). The ultrasound performed at the 30th week of pregnancy described resolution of the thrombosis. The dose of LMWH was then adjusted to a therapeutic dose (enoxaparin 1 mg/kg every 12 hours) and anti-Xa activity was measured to ensure a therapeutic range (0.5–1.2 U/mL). The genetic analysis (detected by nanopore sequencing) showed a heterozygous mutation in c.1366G>C (p.Gly456Arg) in the SERPINC1 gene.
After delivery, she maintained a correct anticoagulation range with VKAs (international normalized ratio [INR] range 2.0–3.0). In October 2021, C1 was diagnosed with ovarian endometrial cancer stage I according to the International Federation of Gynecology and Obstetrics (FIGO). She underwent a hysterectomy and right adnexectomy in January 2022 and required adjuvant chemotherapy with carboplatin, starting in February 2022. The follow-up CT scan revealed cancer remission. She was anticoagulated with LMWH at therapeutic doses (enoxaparin 1 mg/kg every 12 hours) while she received chemotherapy, and this was maintained until complete remission of the cancer. Anti-Xa activity was within the therapeutic range at all times. Twenty-five days after stopping chemotherapy, C1 was admitted to the hospital with moderate exercise-induced dyspnea and dry cough for 7 days. Low-load severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) infection was detected by polymerase chain reaction (PCR). The CT scan showed multiple filling defects related to PE and pneumonia in the right superior lung. The ultrasound of both lower limbs revealed the presence of deep proximal VT in the left leg. No IVC thrombus was shown. The INR at admission was 2.3. LMWH at therapeutic doses (enoxaparin 1 mg/kg every 12 hours) and AT replacement to achieve an AT level between 80% and 120% were administered, reaching a therapeutic anti-Xa activity range. C1 had a good clinical evolution of the pneumonia with supplemental oxygen. She was discharged from the hospital 5 days later; the PCR was then negative for SARS-CoV-2. The INR range was adjusted to 2.5–3.5. Twenty days later, the follow-up ultrasound described the presence of a venous free-floating thrombus in the IVC. No data on IVCA were described. The INR at that moment was 2.8 and the patient remained asymptomatic. The thrombophilia study did not reveal inherited or acquired thrombophilia, except for the previously known AT deficiency. Similarly to the previous in-hospital admission, LMWH at therapeutic doses and AT supplementation were given. Owing to the difficulties in preventing new thrombosis despite correct anticoagulation, an IVC was placed. A new target INR of 3.0–4.0 was established. Three weeks later, the CT scan did not show the free-floating thrombus and the ovarian endometrial cancer remained in radiological and analytical remission. The vena cava filter was then removed. She remains currently asymptomatic and close INR monitoring is performed (INR self-monitoring).
Comments on Case 1
Our patient, in agreement with type I AT deficiency, has similar AT antigen and activity levels. The recurrent thrombotic phenotype, both in the patient and in direct affected relatives, despite correct anticoagulation, also correlates with type I AT deficiency.7,8
Genetic study should be considered in the case of a positive family AT-deficient background at a young age, in all first-degree relatives, even those who are asymptomatic.7,8 A c.1366G>C (p.Gly456Arg) heterozygous mutation in the SERPINC1 gene has been associated with type I AT deficiency.6
Further treatment/prophylactic management is necessary, considering the high thrombotic risk and recurrence rate of inherited AT deficiency.14
Determination of AT activity remains the main diagnostic tool for hereditary AT deficiency. According to the literature, an AT activity below 70% overall, in the context of a positive family thrombotic history, suggests congenital AT deficiency.6 Despite receiving antithrombotic prophylaxis with LMWH, C1 developed her first thrombotic episode during pregnancy, which is a well-known prothrombotic setting.
It has been suggested that warfarin could increase AT levels. However, this is not consistent throughout the literature.21,22 Close INR monitoring is essential to ensure a therapeutic range. However, as reported in C1, despite a correct INR range, thrombosis can appear in the presence of other prothrombotic situations, such as cancer and SARS-CoV-2 infection. Cancer induces a procoagulant state and increases both arterial and venous thrombotic incidence rates (2–5% and 4–20%, respectively). In addition, chemotherapy may decrease anticoagulants and increase procoagulants, such as tissue factor, and can induce platelet activation, all together promoting thrombosis.23 The thrombotic risk could vary depending on the anticancer agent. In particular, C1 received platin-based agents, which are known to increase thrombosis compared to non-platin combinations.24 The mechanism by which platin increases thrombosis is still a matter of debate. In these patients, elevated circulating von Willebrand factor, platelet activation, increased levels of thrombin–antithrombin complexes and increased D-dimer have been described.25,26 Thrombosis and coagulopathy, together with pulmonary injury and inflammation, are clinically associated with coronavirus disease 2019 (COVID-19) infection. The endothelial injury associated with the cytokine storm promotes thrombosis. In addition, low ADAMTS13 activity, probably related to the excess release of von Willebrand factor, contributes to this prothrombotic COVID-19-related state.26
An alternative way to prevent thrombotic recurrence in patients with INR in the therapeutic range could be to increase the optimal INR range or the administration of LMWH with close monitoring of anti-Xa activity, considering, if necessary, AT concentrates to control heparin resistance.27
Our patient developed a new severe thrombotic event despite a proper anticoagulation INR range (INR of 2.8). In that setting, to avoid further episodes, a vena cava filter was recommended. An IVC filter is indicated in patients with VT and contraindication to anticoagulation, failure of anticoagulation or VT progression despite adequate anticoagulation.28 Once the venous free-floating thrombus disappears, the vena cava filter can be removed, and a strict INR control to ensure correct anticoagulation should be performed.
Case 2 (C2)
Massive Cerebral Vein Thrombosis in Childhood
C2 is a 5-year-old girl admitted to a local hospital with sudden tetraparesis with light predominance in her right hemibody and Broca aphasia. Her medical records included an AT deficiency type I (AT activity of 40% and AT antigen of 35%, confirmed in two different samples). A cousin had suffered from cerebral venous thrombosis (CVT) 3 days after birth, her father had developed a deep VT in childhood, and an asymptomatic aunt had been previously diagnosed with inherited AT deficiency. The genetic analysis showed a heterozygous mutation in c.481C>T (p.Arg161Ter) in the SERPINC1 gene.
Hemodynamic stability was achieved during admission to the intensive care unit. An AT activity of 49% was confirmed at admission. A generalized paresthesia, more noticeable in the right upper extremity, was observed. Initial contrast brain CT revealed filling defects affecting the internal cerebral veins, Galeno vein, inferior longitudinal sinus and left transverse sinus, with extension to the left internal jugular vein, suggesting deep CVT. AT replacement to maintain AT levels of 100%, in addition to LMWH (enoxaparin 1 mg/kg twice per day), was immediately initiated. However, the intracranial pressure increased, and clinical worsening was evident, with the presence of seizures and posterior loss of consciousness, probably secondary to obstructive hydrocephalus. Orotracheal intubation was required. Mechanical thrombectomy was then carried out and a partial recanalization was achieved. A 4 mg bolus of local alteplase was injected in the straight sinus, followed by a continuous alteplase infusion at a dose of 1 mg per hour. AT replacement and LMWH 1 mg/kg every 12 hours were administered, reaching AT levels of 100% and an anti-Xa activity in the therapeutic range. The patient experienced a gradual neurological improvement, coinciding with a radiological reduction and posterior resolution of the massive thrombosis and edema. She is currently on anticoagulation (INR target 2.5–3.5) and asymptomatic.
Comments on Case 2
CVT is an uncommon but severe condition, more frequently affecting young children.9 Although high-risk inherited thrombophilia has been related to common sites of thrombosis such as deep veins in the legs, an association with rare presentations has been described.14
More than half of the patients with AT deficiency develop a spontaneous thrombosis.5,8 CVRFs should be controlled to minimize thrombotic risk, and antithrombotic prophylaxis with heparin should be considered in high-risk prothrombotic situations.6 In our patient, there was no trigger associated with thrombosis. Of note, her younger cousin developed a CVT 3 days after birth.
AT replacement may be adequate in an acute VT, especially in unusual severe thrombosis, despite adequate anticoagulation and heparin resistance.6,17 Our patient was, consequently, the optimal candidate for both anticoagulation at therapeutic doses and AT replacement, owing to the severity and atypical presentation.
In CVT, endovascular thrombolysis with or without mechanical thrombectomy may be beneficial in patients with poor prognosis despite anticoagulant treatment with heparin, due mainly to the impossibility of dissolving the extensive thrombus. However, this is based on case series and not on controlled randomized trials.29 Siddiqui et al included a systematic review of 185 patients undergoing mechanical thrombectomy, and a mean recanalization of 95% was obtained. These data were obtained from small retrospective studies.30
The mutation described in C2 and her relatives is associated with hereditary quantitative AT deficiency (type I) and carries a high thrombotic risk.31 In fact, all affected relatives except for one aunt developed thrombosis at a very young age. A peak incidence of thrombosis, especially CVT during the neonatal period, has been described in hereditary AT-deficient patients.32 The physiological AT deficiency at birth is exacerbated by the hereditary defect of AT, inducing prothrombotic situations such as hypoxia and the release of tissue factor more easily, together with possible trauma and manipulation during delivery.32
Case 3 (C3)
Female Adult with Neither Familiar nor Personal Thrombosis and Congenital AT Deficiency
C3 is a 47-year-old businesswoman, a habitual smoker of 10 cigarettes per day for 15 years, with no other additional CVRFs. No family history of thrombosis was reported. In April 2001, in the setting of three miscarriages before the 10th week of pregnancy, a thrombophilia study was performed. A type II AT deficiency was detected, confirmed in two different samples (AT activity of 46%, AT antigen of 80%). Family screening was completed, and her father, uncle and grandfather had similar AT levels (activity and antigen) to C3. The genetic analysis showed a heterozygous mutation in c.235C>T (p.Arg79Cys) in the SERPINC1 gene. None of them had previously developed thrombosis. C3 is currently taking hormonal replacement with transdermal estrogen owing to early menopause.
Comments on Case 3
Despite the high thrombotic risk associated with hereditary AT deficiency, there may be adult patients with known underlying AT deficiency and no previous thrombotic events. This uncommon scenario could happen mainly in patients with no CVRFs and a healthy lifestyle. Up to 40% of patients with hereditary AT deficiency had thrombosis associated with a transient risk factor.6 C3, except for smoking, has an active healthy lifestyle, as do her first-degree affected relatives. They should be given prophylactic heparin during a prothrombotic situation, such as immobilization, pregnancy or active cancer. Hormonal treatment containing a high estrogen dose is not recommended, as it could increase the thrombotic risk. Low-dose estrogen pills, pills containing only progestagen or an intrauterine device are treatments of choice in high-risk prothrombotic women who require hormonal treatment.32 In postmenopausal women, transdermal estrogen does not increase thrombotic risk.33 The most suitable option should be carefully considered by the gynecologist, depending on the reason for hormonal treatment and the presence of other CVRFs.32,33
The genetic mutation could play a role in the severity of thrombosis. The heterozygous missense mutation, a cytosine to thymine substitution at nucleotide position 235 in exon 2 of the SERPINC1 gene (p.Arg79Cys), has been associated with a low prevalence of thrombosis. This is a mutation in the heparin binding site, described in type IIb AT deficiency. Subjects with this mutation, mainly heterozygous patients, rarely develop thrombosis.32 Alhenc-Gelas et al did not describe an increase in the endogenous thrombin potential in type IIb heterozygous patients compared to a control group without AT deficiency (2142 nMxmin vs 2299 nMxmin), being associated with a low risk of VT.34 Yoo et al reported the first patient with this heterozygous mutation who had deep VT and a family history of thrombosis.35
In case antithrombotic therapy with heparin is needed, patients with heparin binding site mutation may require AT replacement, as heparin itself could not generate an appropriate antithrombotic effect.6,17
Case 4 (C4)
Adult Man with Recurrent Thrombosis Despite Correct Anticoagulation
C4 is a 64-year-old man who requires indefinite anticoagulation owing to recurrent PE. No family history of thrombophilia was reported. The first PE and deep VT in his right leg occurred in 2000 spontaneously at the age of 46. He had, at that time, several CVRFs, including hypertension, dyslipidemia and obesity (body mass index of 33 kg/m2), and he had been a smoker of 15 cigarettes per day for 15 years. He was being followed by the general practitioner owing to hepatic steatosis. Anticoagulation with VKAs was then initiated, reaching the INR target of 2.5 easily. The CVRFs significantly improved as a result of a healthy lifestyle and proper medication for hypertension and dyslipidemia. An AT activity of 65% was detected, without any other thrombophilia defects. This mild AT deficiency was not considered of relevance and was first reported in the low range of normality (in our laboratory, the normal levels for AT activity range between 70% and 100%). However, in further routine analytical controls, AT activity remained between 60% and 65% and the AT antigen was 53%, confirmed in two different samples. Therefore, in the setting of an idiopathic thrombosis and the previous analytical abnormalities, AT deficiency type I was confirmed. The patient had a heterozygous point mutation in the SERPINC1 gene, c.1219–3C>A, affecting intron 6, very close to the exon 6 donor sequence (IVS6-3C>A). Four years later, the patient was admitted to the emergency department with idiopathic recurrent PE despite a correct INR (2.4). He had a favorable clinical evolution with LMWH at therapeutic doses (enoxaparin 1 mg/kg every 12 hours) and AT concentrates to reach an AT activity of 100%. At hospital discharge, the INR range was adjusted to 2.5–3.5. No further thrombotic events have been reported.
Comments on Case 4
Similarly to C3, in a few families AT deficiency is not diagnosed at an early age because of the absence of personal or family thrombotic history. C4 developed his first thrombotic event at the age of 46 in the setting of different CVRFs. The mild AT activity deficiency was first considered to lie in the range of normality. This result correlated with the diagnosis of hepatic steatosis included in his medical records. However, the rest of the proteins of hepatic origin included in the thrombophilia study, such as protein S, protein C and plasminogen, were strictly normal, as was the hepatic function profile (transaminases and bilirubin). Later, once AT type I deficiency had been confirmed, the genetic family study was performed. His older sister did not carry the mutation and had a normal AT activity (90%), while his parents were not available for the genetic study. C4 has been on anticoagulation since this first episode owing to high recurrent thrombotic rate.16 Strict INR monitoring is essential to prevent further thrombotic episodes, ensuring no INR below the target range. In addition, appropriate control of CVRFs is necessary to reduce the thrombotic risk. C4 achieved a substantial improvement in his CVRFs with medication for hypertension and dyslipidemia, and having lost weight. An appropriate level of anticoagulation was achieved, with no INR fluctuations. However, he developed a new thrombosis that required INR target adjustment.
The mutation detected in the SERPINC1 gene could affect the appropriate processing of exon/intron 6 during mRNA formation, and it is associated with high thrombotic risk.36 This mutation has been described previously.36,37
Conclusions
- Inherited AT deficiency is the most severe type of thrombophilia, with a remarkable clinical heterogeneity that requires indefinite anticoagulation.
- Genetic testing should be considered in all first-degree relatives at a young age, and appropriate treatment/prophylactic management is necessary to minimize thrombotic risk.
- Type IIb confers a lower thrombotic risk and may be underdiagnosed.
Ethics and Consent
The study participants have given written informed consent to participate and for publication of the data. Parental consent was obtained for the case of a minor. Institutional approval was not required for publication.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rosenberg RD, Damus PS. The purification and mechanism of action of human antithrombin-heparin cofactor. J Biol Chem. 1973;248(18):6490–6505. doi:10.1016/S0021-9258(19)43472-8
2. Egeberg O. Inherited antithrombin deficiency causing thrombophilia. Thromb Diath Haemorrh. 1965;13:516–530.
3. Tsuchida T, Hayakawa M, Kawahara S, Kumano O. Thrombin generation capacity is enhanced by low antithrombin activity and depends on the activity of the related coagulation factors. Thromb J. 2022;20(1). doi:10.1186/s12959-022-00388-w
4. Colucci G, Tsakiris D. Thrombophilia screening revisited: an issue of personalized medicine. J Thromb Thrombolysis. 2020;49(4):618–629. doi:10.1007/s11239-020-02090-y
5. Martinelli I, Mannucci PM, De Stefano V, et al. Different risks of thrombosis in four coagulation defects associated with inherited thrombophilia: a study of 150 families. Blood. 1998;92(7):2353–2358. doi:10.1182/blood.V92.7.2353
6. Pabinger I, Thaler J. How I treat patients with hereditary antithrombin deficiency. Blood. 2019;134(26):2346–2353. doi:10.1182/blood.2019002927
7. Bravo-Pérez C, Toderici M, Chambers JE, et al. Full-length antithrombin frameshift variant with aberrant C-terminus causes endoplasmic reticulum retention with a dominant-negative effect. JCI Insight. 2022;7(19):e161430. doi:10.1172/jci.insight.161430
8. Bravo-Perez C, De la Morena Barrio ME, Vicente V, Corral J. Antithrombin deficiency as a still underdiagnosed type of thrombophilia: a primer for internists. Pol Arch Intern Med. 2020;130(10):868–877. doi:10.20452/pamw.15371
9. Patnaik MM, Moll S. Inherited antithrombin deficiency: a review. Haemophilia. 2008;14(6):1229–1239. doi:10.1111/j.1365-2516.2008.01830.x
10. De la Morena‑Barrio ME, Martinez‑Martinez I, de Cos C, et al. Hypoglycosylation is a common finding in antithrombin deficiency in the absence of a SERPINC1 gene defect. J Thromb Haemost. 2016;14(8):1549–1560. doi:10.1111/jth.13372
11. De la Morena-Barrio ME, Gindele R, Bravo-Pérez C, et al. High penetrance of inferior vena cava system atresia in severe thrombophilia caused by homozygous antithrombin Budapest 3 variant: description of new syndrome. Am J Hematol. 2021;96(11):1363–1373. doi:10.1002/ajh.26304
12. Muller-Knapp M, Classen CF, Knofler R, et al. Coexistence of antithrombin deficiency and suspected inferior vena cava atresia in an adolescent and his mother: case report and clinical implications. Thromb J. 2021;19:105–112. doi:10.1186/s12959-021-00360-0
13. Lukaseder J, Feldman R, Steiner A. Recurrent vein thrombosis with agenesis of the inferior vena cava and AT III deficiency. Phlebologie. 2017;46(01):31–33. doi:10.12687/phleb2345-1-2017
14. De la Morena‑Barrio B, Orlando C, de la Morena‑Barrio ME, Vicente V, Jochmans K, Corral J. Incidence and features of thrombosis in children with inherited antithrombin deficiency. Haematologica. 2019;104(12):2512–2518. doi:10.3324/haematol.2018.210666
15. Di Minno MND, Ambrosino P, Ageno W, Rosendaal F, Di Minno G, Dentali F. Natural anticoagulants deficiency and the risk of venous thromboembolism: a meta‑analysis of observational studies. Thromb Res. 2015;135(5):923–932. doi:10.1016/j.thromres.2015.03.010
16. Croles FN, Borjas‑Howard J, Nasserinejad K, Leebeek FW, Meijer K. Risk of venous thrombosis in antithrombin deficiency: a systematic review and Bayesian meta‑analysis. Semin Thromb Hemost. 2018;44:315–326. doi:10.1055/s-0038-1625983
17. Bauer KA, Nguyen-Cao TM, Spears JB. Issues in the diagnosis and management of hereditary antithrombin deficiency. Ann Pharmacother. 2016;50(9):758–767. doi:10.1177/1060028016651276
18. Elsebaie MAT, van Es N, Langston A, Büller HR, Gaddh M. Direct oral anticoagulants in patients with venous thromboembolism and thrombophilia: a systematic review and meta-analysis. J Thromb Haemost. 2019;17(4):645–656. doi:10.1111/jth.14398
19. Soerajja Bhoelan B, Mulder R, Lukens MV, Meijer K. Direct oral anticoagulants in antithrombin deficiency: initial experience in a single center. Thromb Haemost. 2021;121(02):242–245. doi:10.1055/s-0040-1715647
20. Campello E, Spiezia L, Simion C, et al. Direct oral anticoagulants in patients with inherited thrombophilia and venous thromboembolism: a prospective study. J Am Heart Assoc. 2020;9(23):e018917. doi:10.1161/JAHA.120.018917
21. O’Brien JR, Etherington MD. Effect of heparin and warfarin on antithrombin III. Lancet. 1977;2(8050):1231.
22. Sanfelippo MJ, Engel JM, Onitilo AA. Antithrombin levels are unaffected by warfarin. Arch Pathol Lab Med. 2014;138(7):967–968. doi:10.5858/arpa.2013-0065-OA
23. Grover SP, Hisada YM, Kasthuri RS, Reeves BN, Mackman N. Cancer therapy-associated thrombosis. Arterioscler Thromb Vasc Biol. 2021;41(4):1291–1305. doi:10.1161/ATVBAHA.120.314378
24. Moik F, van Es N, Posch F, et al. Gemcitabine and platinum-based agents for the prediction of cancer associated venous thromboembolism: results from the Vienna cancer and thrombosis study. Cancers. 2020;12(9):2493. doi:10.3390/cancers12092493
25. Seng S, Liu Z, Chiu SK, et al. Risk of venous thromboembolism in patients with cancer treated with Cisplatin: a systematic review and meta-analysis. J Clin Oncol. 2012;30(35):4416–4426. doi:10.1200/JCO.2012.42.4358
26. Marco A, Marco P. Von Willebrand factor and ADAMTS13 as clinical severity markers in patients with COVID-19. J Thromb Thrombolysis. 2021;52(2):497–503. doi:10.1007/s11239-021-02457-9
27. Luxembourg B, Pavlova A, Geisen C, et al. Impact of the type of SERPINC1 mutation and subtype of antithrombin deficiency on the thrombotic phenotype in hereditary antithrombin deficiency. Thromb Haemost. 2014;111(2):249–257. doi:10.1160/TH13-05-0402
28. Li X, Haddadin I, McLennan G, et al. Inferior vena cava filter: comprehensive overview of current indications, techniques, complications and retrieval rates. Vasa. 2020;49(6):449–462. doi:10.1024/0301-1526/a000887
29. Ferro JM, Bousser GM, Canhao P, et al. European Stroke Organization Guidelines for the diagnosis and treatment of cerebral venous thrombosis. Europ Stroke Journal. 2017;2(3):195–221. doi:10.1177/2396987317719364
30. Siddiqui FM, Dandapat S, Banerjee C, et al. Mechanical thrombectomy in cerebral venous thrombosis: systematic review of 185 cases. Stroke. 2015;46(5):1263–1268. doi:10.1161/STROKEAHA.114.007465
31. Gandrille S, Vidaud D, Emmerich J, et al. Molecular basis for hereditary antithrombin III quantitative deficiencies: a stop codon in exon IIIa and a frameshift in exon VI. Br J Haematol. 1991;78(3):414–420. doi:10.1111/j.1365-2141.1991.tb04457.x
32. Abou-Ismail MY, Sridhar DC, Nayak L. Estrogen and thrombosis: a bench to bedside review. Thromb Res. 2020;192:40–51. doi:10.1016/j.thromres.2020.05.008
33. Canonico M, Oger E, Plu-Bureau G, et al. Hormone therapy and venous thromboembolism among postmenopausal women. Circulation. 2007;115(7):840–845. doi:10.1161/CIRCULATIONAHA.106.642280
34. Alhenc-Gelas M, Canonico M, Picard V. Influence of natural SERPINC1 mutations on ex vivo thrombin generation. J Thromb Haemost. 2010;8(4):845–848. doi:10.1111/j.1538-7836.2010.03750.x
35. Yoo JH, Maeng HY, Kim HJ, Lee KA, Choi JR, Song J. A heparin binding site Arg79Cys missense mutation in the SERPINC1 gene in a Korean patient with hereditary antithrombin deficiency. Ann Clin Laboratory Sci. 2011;41:89–92.
36. De la Morena Barrio ME, López-Gálvez R, Martínez-Martínez I, et al. Defects of splicing in antithrombin deficiency. Res Pract Thromb Haemost. 2017;1(2):216–222. doi:10.1002/rth2.12025
37. Human Gene Mutation Database (HGMD) [homepage on the Internet]; 2023. Available from: www.hgmd.cf.ac.uk.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.