")
Back to Journals » Journal of Blood Medicine » Volume 14
Current Status and Challenges in Delivering Comprehensive Care for Patients with Hemophilia
Authors Nomura S
Received 23 October 2023
Accepted for publication 11 December 2023
Published 15 December 2023 Volume 2023:14 Pages 629—637
DOI https://doi.org/10.2147/JBM.S446204
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Shosaku Nomura
Center of Thrombosis and Hemostasis, Kansai Medical University Medical Center, Moriguchi, Osaka, Japan
Correspondence: Shosaku Nomura, Center of Thrombosis and Hemostasis, Kansai Medical University Medical Center, 10-15 Fumizono-cho, Moriguchi, Osaka, 570-8507, Japan, Tel + 81 6 6992 1001, Fax + 81 6 6992 1066, Email [email protected]
Abstract: The importance of comprehensive care as a treatment strategy for patients with hemophilia is recognized worldwide. Comprehensive care entails addressing full spectrum of medical and psychological aspects impacting both patients and their families. The primary objective of comprehensive care for individuals with hemophilia is to enable them to lead their daily lives just as anyone else would. To achieve this goal, it is necessary to have a positive and collaborative approach across various healthcare disciplines. This extends beyond clinical specialists, encompassing pediatricians, hematologists, orthopedic surgeons, dental and oral surgeons, gynecologists, nurses, physical therapists, clinical psychologists, and other professionals from diverse fields. This review article discusses the current status and challenges associated with comprehensive care for patients with hemophilia. We categorize these challenges as follows: hemophilic arthritis, rehabilitation, oral care, transitioning from pediatric to adult care, addressing carrier issues, and providing psychological care. There is still substantial work to be undertaken in addressing these hurdles and advancing the quality of comprehensive care for hemophilia patients.
Keywords: hemophilia, comprehensive care, collaborative approach, psychological care, carrier issues
Introduction
Hemophilia A and B are congenital bleeding disorders characterized by abnormalities in coagulation factors (F) VIII and IX, resulting in impaired coagulation factor production and a heightened propensity for bleeding.1,2 Patients with severe phenotypes produce less than 1% (1 IU/dL) of FVIII or FIX activity, experiencing recurrent musculoskeletal, soft tissue, and intracranial hemorrhages, which can sometimes escalate into life-threatening events.3 Moderately affected individuals exhibit residual coagulation factor levels between 1–5% of normal, and spontaneous bleeding occurrence is rare; however, there is a risk of bleeding due to trauma. Conversely, in patients with mild hemophilia with coagulation factor activity of 6% or more, bleeding is usually limited to trauma. Notably, even those with mild hemophilia face a higher risk of intracranial hemorrhage-related mortality compared to individuals without hemophilia.4
At present, the established standard of care for patients with severe hemophilia revolves around maintaining normal FVIII or FIX activity levels factor levels at or above 1%. This objective is achieved through the regular prophylactic administration of factor concentrates from an early age, effectively preventing arthropathy and normalizing life expectancy.5,6 Aditionally, extended half-life formulations have been developed to reduce the frequency of concentrated dosing.7,8 Furthermore, the introduction of innovative therapies, such as emicizumab, has been transformative in the clinical management of hemophilia. This therapy not only improve compliance with prophylaxis but also provides alternative treatments for patients with inhibitors.9,10 Finally, progress is being made toward gene therapy.11,12 Clinical trials for gene replacement therapy are actively developing.13
The significance of comprehensive care as a treatment strategy for patients with hemophilia is recognized worldwide. Comprehensive care extends to the thorough management of all medical and psychological aspects impacting patients and their families.14 To provide optimal care for patients with hemophilia, it is important to understand and recognize the various factors involved.15 The evolution of comprehensive clinical management for hemophilia has given rise to the establishment of hemophilia treatment centers (HTCs).16 The core team members of the HTC include not only hematologists and clinical specialists, but also nurses, physical therapists, clinical psychologists, and professionals from various fields.17,18 This review aims to discuss the current status and challenges encountered in the realm of comprehensive care for hemophilia patients. In preparing the text, we searched for articles published in PuBMed over the past 10 years using the keywords “hemophilia”, “comprehensive care”, and “review”, and selected five articles to serve as reference materials.19–23
Establishment of Comprehensive Care and Its Objectives and Methods
The primary aim of comprehensive care is to enable hemophilia patients to lead their daily lives with the same nor, alcy as anyone else. To fulfill the aforementioned objectives of comprehensive care, it is imperative to involve not only clinical specialists such as pediatricians, hematologists, orthopedists, dental and oral surgeons, and gynecologists but also enlist the active participation of nurses, physical therapists, clinical psychologists, and various other professionals from their respective domains (Figure 1). Specifically, the following intensive efforts are needed.
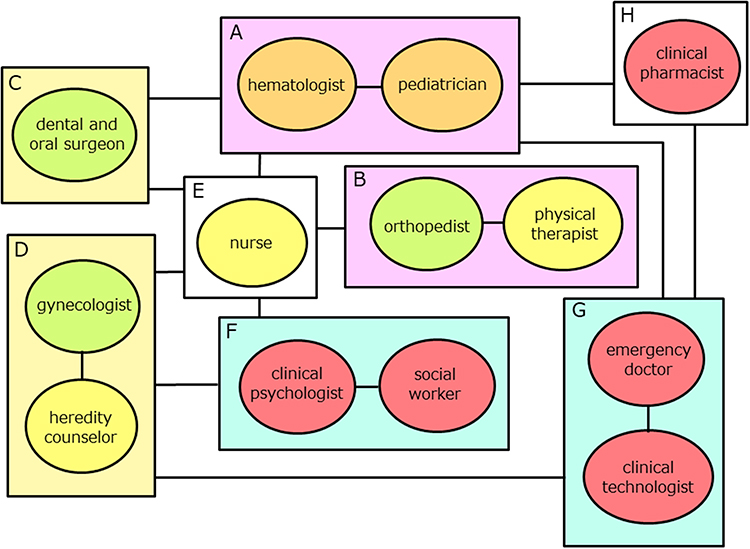 |
Figure 1 Components of Comprehensive Hemophilia Care. (A) Hematologists and pediatricians play a fundamental role in comprehensive care and complete transitional care; (B) Orthopedist and physical therapist are responsible for joint evaluation and treatment; (C) Dental and oral surgeons are responsible for oral care; (D) Gynecologists and heredity counselors are involved in the care and education of carriers; (E) Nurses are responsible for mental health care as well as assisting in the treatment of each department; (F) Clinical psychologists and social workers are responsible for mental health care as well as supporting social welfare; (G) Emergency doctors and clinical technologist are responsible for the care of patients in the event of an emergency. In addition, the clinical technologist contributes to the medical assistance of each department; (H) Clinical pharmacists provide information on therapeutic drugs to the departments and manage drug inventories. |
Foster Intra-Hospital Cooperation and Enhance Medical Care
- Conduct joint evaluations at an early age (Figure 1A–C).
- Promptly formulate appropriate treatment plans for patients with moderate disease (Figure 1A and B).
- Conduct joint consultations between pediatrics and hematology, with an eye towards transitions for patients approaching adulthood (Figure 1A).
- Provide care for carriers of the disease (Figure 1A and D–F).
Train Medical Personnel
- Advocate the importance of cooperation among medical departments (Figure 1A).
- Utilize the center for practical training of students to raise awareness of the disease.
- Offer specialized training for hemophiliacs in pediatrics and internal medicine, as needed (Figure 1A).
Conduct Public Relations Activities Outside the Hospital
- Disseminate information regarding the comprehensive care system on the medical institution’s website.
- Extend the scope of comprehensive care to encompass patients in nearby areas.
Hemophilic Arthropathy
Hemophilic arthropathy is caused by repeated intra-articular hemorrhages, with iron deposits in the joints being recognized as the primary cause of this condition.24 Hemophilic arthropathy can manifest in any major joint, such as the shoulder, elbow, hand, hip, knee, and foot, with the elbow, knee, and ankle joints referred to as index joints, being particularly susceptible.25,26 Joints that undergo three or more non-traumatic hemorrhages within a six-month period are called target joints.27 In essence, alongside the six index joints, these target joints are extremely important in the evaluation of hemophilic arthropathy.
When a joint experiences bleeding, phagocytic cells in the synovial membrane are triggered into action to remove blood components stored within the joint cavity. This process leads to the development of synovitis.25 According to the guidelines of the World Hemophilia Federation (WHF), chronic synovitis is defined as the persistence of synovitis for over three months.28 MRI studies examining joint changes over time generally reveal that soft tissue changes can be reversed, whereas alterations in bone and cartilage are irreversible.29 Contemporary suggestions recommend the use of refractory synovitis to describe irreversible synovitis that persists for more than three months.30 Therefore, the early diagnosis and appropriate treatment of synovitis are considered important for preventing the progression of arthropathy. Traditionally, the management of hemophilia has centered around patients complaints of bleeding symptoms. However, relying solely on patient-reported symptoms makes it challenging to identify all instances of bleeding. In the case of micro-hemorrhages without clinical signs, lesions may be identified for the first time through MRI scans in joints exhibiting no subjective symptoms.5 Several reports presented in recent years highlight the discrepancy between the patients’ self-reported symptoms and the extent of arthropathy.31–33 In essence, it has become evident that the diagnosis of hemorrhage and the severity of arthropathy cannot be determined solely based on patient complaints, necessitating an objective test for evaluating hemorrhage and synovitis.
To prevent and manage hemophilic arthropathy effectively, it is crucial to diagnose arthropathy and devise a treatment plan before irreversible bone and cartilage changes occur. As hemorrhage and synovitis changes may manifest as early as childhood, pediatricians must identify joint changes in infants and young children as early as possible. In this context, joint echocardiography is superior to MRI as an imaging evaluation method that simultaneously caters to this target population and purpose, making it particularly valuable for patients with hemophilia.34 In comprehensive care, simplicity and speed are considered key factors in selecting a joint echocardiographic technique for assessing hemophilia arthropathy. One such method well suited for this task is Hemophilia Early Arthropathy Detection with Urtrasound (HEAD-US), which has gained popularity.35 Originally designed for outpatient use, this method provides a simple and swift means of detecting early hemophilic arthropathic changes. Research indicates a correlation between the HEAD-US score and the Hemophilia Joint Health Score (HJHS).36 However, some reports have noted limitations in the identification of soft tissue characteristics depicted through arthrography.37 To address this limitation, the Joint Tissue Activity and Damage Examination (JADE) has been newly proposed, allowing the measurement of soft tissue changes and bone and cartilage thickness.38 Accurate recognition of the hemophilic arthropathy status is crucial in determining the course of treatment. While a definitive diagnosis by an orthopedic surgeon remains vital, an initial examination conducted by a pediatrician or internist using joint echocardiography in the routine care setting is also indispensable within the scope of comprehensive care (Figure 1A and B).
Rehabilitation
The most prevalent musculoskeletal observations in patients with hemophilia are blood specimens affecting both muscles and joints.39,40 Pediatricians or hematologists are always prepared to address the acute manifestations of hemophilia.18,28 Nevertheless, they may not always be adequately equipped to manage the chronic phase of hemophilia and prevent the loss of functional independence. In this context, it is important to underscore the indispensable role that physical therapists play in hemophilia care (Figure 1B).41,42
Treatment and rehabilitation should not be limited solely to patients with established arthropathy but should extend to those encountering unexpected major bleeding events or postoperative musculoskeletal conditions. These scenarios benefit from comprehensive evaluation by a physical therapist working in concert with the hemophiliac and orthopedic surgeon.43 Furthermore, the role of the physical therapist extends to educating both the patient and their parents regarding non-hematologic strategies for addressing bleeding episodes. This includes offering recommendations such as orthotic prescriptions to facilitate the patient’s continued physical activity and participation in sports.44 Initially, the core objective of physical therapy was to address an individuals exercise requirement and potential.45 Notably, guidelines for hemophilia treatment advocate for a diverse array of services, including physical therapy offering specifically tailored for hemophilia patients.46,47 Successful treatment of hemophilia requires collaboration between both hemophiliacs and physical therapists. Particularly, the assessment of the musculoskeletal system by a physical therapist can significantly enhance the quality of therapeutic care.48
In recent years, remarkable advancement have been made in hemophilia bleeding control, with the achievement of hemostasis now considered a matter of course.7,10,49 Moreover, the rapid progression of therapeutic modalities aimed at curing hemophilia, such as gene therapy, is undeniable promising.11–13,50,51 These latest advances in hemophilia treatment have had a major impact on the development of comprehensive care strategies. However, the need for appropriate rehabilitation by skilled physical therapists remains (Figure 1B).52
Oral Care
Oral healthcare encompasses a comprehensive approach aimed at preserving the well-being of the oral cavity, its adjacent structures, and associated organs.53 Oral care is extremely important in maintaining fundamental oral functions such as eating, chewing, swallowing, and articulation.54 In the context of hemophilia, special measures are required for dental care due to the heightened the risk of bleeding.55 Many individuals with hemophilia tend to postpone dental visits, even when displaying symptoms of tooth decay or periodontal disease, owing to the fear of triggering a bleeding episode. Oral bleeding incidents in individuals with hemophilia are more prone to occur when they experience falls that impact their mouth or unintentionally bite oral soft tissue.55,56 Infants face an additional risk associated with tooth eruption.57 Conversely, tooth decay and periodontal disease, if severe and untreated, are not infrequent causes of irreversible bleeding.58 Properly maintained gums are less susceptible to bleeding during activities such as brushing. Thus, individuals with hemophilia should become familiar with daily oral care practices.59
To provide effective dental care to patients with hemophilia, it is important that they possess a solid awareness of their dental health condition. This, in turn, necessitates instruction in proper brushing techniques and education on effective preventive dental care methods.60 A comprehensive hemophilia practice can accomplish these goals (Figure 1A, C and E). Close collaboration among the patient, their family, and healthcare professionals can lead to successful oral healthcare management.61 It is evident that individuals with hemophilia experience a lower quality of life with respect to oral health compared to their healthy peers.59 Therefore, it is strongly advised that individuals with hemophilia receive routine preventive dental care at a hospital or clinic. They should undergo check-ups every six months, and the frequency may need to be increased based on their dental health status. In this context, both visual examination and imaging techniques can be valuable.62 Regular oral care is recommended to improve the quality of life for individuals with hemophilia.
Transition
The transition from pediatric to adult care has garnered significant attention across various medical conditions. It is crucial to acknowledge that environmental factors surrounding a disease can significantly differ across various ages and stages of development. Although the exact timing of the transition from childhood to adulthood remains uncertain, adolescence serves as a pivotal indicator.63 Thus, comprehensive hemophilia care should commence in early adolescence with a conscientious approach to developmentally sensitive care transitions in mind.64
The challenges encountered during the transition from childhood to adulthood are distinct in many respects. Additionally, concurrent medical issues can often complicate the journey into adulthood. In essence, the elements that define this transition should be recognized as unique to pediatric and adult care (Figure 1A).65 The transition of care is a critical period for young individuals with hemophilia, and every effort must be made to ensure it does not lead to a decline in their quality of life (QOL).
Young adults with hemophilia face unique challenges during the transition to adulthood. These include issues associated with switching healthcare systems, cultivating mature interpersonal relationships, and establishing independent careers. Furthermore, recent years have witnessed the use of safer factor products during childhood, resulting in fewer joint problems and lower rates of HIV transmission, all of which may be significantly different from the experiences of older patients with hemophilia.66 To avoid complications following the transition, it is imperative for patients, their families, and healthcare professionals to be fully aware of the fundamental prerequisites for a successful transition from pediatric to adult care (Table 1). It is also imperative to fully recognize the importance of facilitating the transfer of hemophilia patients and establishing suitable transition programs to cater to the needs of the participants.64,67
 |
Table 1 Items to Be Addressed for Transition |
Carrier
Because of the X-linked inheritance of hemophilia, it is a common belief that the condition primarily affects males, with female carriers assumed to be asymptomatic. However, it is important to note that female carriers also exhibit low coagulation factor activity levels, which can result in abnormal bleeding. In some instances, these cases should be classified as hemophilia.68 A previous cross-sectional observational study of female carriers found reduced coagulation factor levels in 23% of eligible cases, confirming an association with bleeding symptoms.69 In such cases, invasive procedures should be conducted after optimizing hemostasis, as in patients with mild hemophilia.70
Most potential carriers comprise adolescent and young adult women who have yet to experience childbirth and consequently may lack knowledge regarding the risks of being carriers. Therefore, presumptive carriers should be identified through genetic counseling starting around the age of 16.70 In the course of counseling, they should receive comprehensive information and active education regarding their reproductive choices, including options such as acceptance of risk, prenatal diagnosis, and preimplantation genetic diagnosis (Figure 1D).71 In the absence of specific bleeding symptoms, carriers may be unaware of their condition. Thus, participation in comprehensive care can be instrumental in making them aware of their susceptibility to bleeding disorders.
The methods for identifying carriers of hemophilia initially relied on phenotyping based on coagulation tests and have more recently evolved to genetic linkage determinations and next-generation sequencing studies that offer more accurate results.72,73 Similarly, methods for prenatal diagnosis of hemophilia have advanced from testing FVIII levels in fetal blood samples at 20–22 weeks of gestation to genetic analysis of fetal DNA extracted from chorionic tissue at 11–14 weeks gestation.73 These advancements may also be crucial in enhancing our understanding of carriers within the context of comprehensive hemophilia care.
Mental Health Care
Individuals with hemophilia and their parents grapple with mental stress and anxieties at various stages of the disease.74 One psychological challenge encountered by patients at an early age is the perception of a disparity with healthy peers with regard to exercise. As long as the risk of bleeding is a concern, exercise restrictions are often inevitable.75 However, during this period, these concerns can be more pronounced in the parents than in the patients themselves,76 raising various anxieties, from immediate situations to the future, which can significantly affect their marital relationships.74 Clinical psychologists, social workers, and nurses play a significant role in this regard (Figure 1E and F).
Adolescent individuals with hemophilia share the same concerns as their healthy peers of the same age, such as club activities, romantic relationships, and aspirations for higher education and employment.77 Many of these concerns are difficult to discuss with relatives, and a clinical psychologist is expected to come into play. Because of advances in coagulation factor replacement therapy, these patients are often more active than one might expect.78 In adolescence, their energy levels surge, accompanied by increased muscular strength, potentially leading to a temporary oversight of their condition, even though it has not been cured. Nevertheless, it is crucial for them to maintain full awareness joint conditions might deteriorate, even without visible swelling, necessitating periodic radiographic assessments.31,32 In the transition from childhood to adolescence, it is also important to pass on the essential skill of securing the route of administration of therapeutic preparations (Figure 1A). In adult cases, parental assistance in securing the route should no longer be anticipated, thereby amplifying the role of skilled nurses in providing technical guidance (Figure 1E).
Other
Understanding the characteristics of the drug and the frequency of side effects is important in deciding on a therapeutic regimen for a hemophiliac, and checking the availability of the drug at the facility should not be ignored. Explanation of specific coagulation abnormalities to the carrier and their reliability must also not be forgotten (Figure 1D). Furthermore, there are many important issues to be addressed, such as manuals for emergency transport in the event of a sudden change in a patient, confirmation of the presence or absence of inhibitors, and means of obtaining the drugs used (Figure 1A, H and G). After all, it should be known that collaboration among many specialists is the cornerstone of comprehensive care.
Conclusions
The success of comprehensive hemophilia care hinges on the collaborative team approach adopted by the various healthcare professionals involved in the patient’s care. Advances in hemophilia-related treatment methods and the constructive contributions from numerous healthcare professionals have led to significant changes in comprehensive care, compared to over three decades ago. Nonetheless, these advancements have also unveiled new challenges.79,80 For example, the availability of innovative therapies must be available for comprehensive care centers to maintain high quality care, and the addition of clinical pharmacists to the comprehensive care model for hemophilia must lead to improved patient outcomes and savings in drug costs. In addition, disparities in the healthcare systems across different countries present a major challenge, and treatments suitable for patients have not necessarily reached all populations. Regarding the management of carriers, current efforts may only be scratching the surface. Thus, much work remains to surmount the challenges confronting comprehensive care.
Acknowledgments
The author thanks Ryosuke Matsuno, Yukihiro Noda, Taichi Omachi, Takashi Yamazoe, Minoru Murata, Fumito Kobayashi, Yuka Kojima, Shunsuke Sawada, Aya Yoshida, Akio Kamiya, Meguru Taguchi, Shun Ito, Reimi Matsuda, Akiko Matsumoto, Chika Sato and Saki Shimada, participating members of the Hemophilia Comprehensive Care at Kansai Medical University.
Disclosure
S. Nomura: Received honoraria for participation in lectures from Chugai, Novo Nordisk, Sanofi, CSL Behring, Takeda, and Japan Blood Products Organization. The author reports no other conflicts of interest in this work.
References
1. Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature. 1984;312(5992):342–347. doi:10.1038/312342a0
2. Camerino G, Grzeschik KH, Jaye M, et al. Regional localization on the human X chromosome and polymorphism of the coagulation factor IX gene (hemophilia B locus). Proc Natl Acad Sci USA. 1984;81(2):498–502. doi:10.1073/pnas.81.2.498
3. Nathwani AC, Tuddenham EG. Epidemiology of coagulation disorders. Baillieres Clin Haematol. 1992;5(2):383–439. doi:10.1016/s0950-3536(11)80025-9
4. Loomans JI, Fijnvandraat K. Mortality caused by intracranial bleeding in non-severe hemophilia a patients: reply. J Thromb Haemost. 2017;15(8):1710–1711. doi:10.1111/jth.13756
5. Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535–544. doi:10.1056/NEJMoa067659
6. Darby SC, Kan SW, Spooner RJ, et al. Mortality rates, life expectancy, and causes of death in people with hemophilia A or B in the United Kingdom who were not infected with HIV. Blood. 2007;110(3):815–825. doi:10.1182/blood-2006-10-050435
7. Lambert T, Benson G, Dolan G, et al. Practical aspects of extended half-life products for the treatment of haemophilia. Ther Adv Hematol. 2018;9(9):295–308. doi:10.1177/2040620718796429
8. Nogami K, Shima M, Fukutake K, et al. Correction to: efficacy and safety of full-length pegylated recombinant factor VIII with extended half-life in previously treated patients with hemophilia A: comparison of data between the general and Japanese study populations. Int J Hematol. 2018;107(1):123–124. doi:10.1007/s12185-017-2369-z
9. Shima M, Hanabusa H, Taki M, et al. Factor VIII-mimetic function of humanized bispecific antibody in hemophilia A. N. Engl J Med. 2016;374(21):2044–2053. doi:10.1056/NEJMoa1511769
10. Oldenburg J, Levy GG. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. 2017;377(22):2194–2195. doi:10.1056/NEJMc1712683
11. Kaminski TW, Ju EM, Gudapati S, et al. Defenestrated endothelium delays liver-directed gene transfer in hemophilia A mice. Blood Adv. 2022;6(12):3729–3734. doi:10.1182/bloodadvances.2021006388
12. Rasul E, Hallock R, Hellmann M, et al. Gene therapy in hemophilia: a transformational patient experience. J Patient Exp. 2023;10:23743735231193573. doi:10.1177/23743735231193573
13. Pipe SW, Gonen-Yaacovi G, Segurado OG. Hemophilia A gene therapy: current and next-generation approaches. Expert Opin Biol Ther. 2022;22(9):1099–1115. doi:10.1080/14712598.2022.2002842
14. Evatt BL, Black C, Batorova A, Street A, Srivastava A. Comprehensive care for haemophilia around the world. Haemophilia. 2004;10(Suppl s4):9–13. doi:10.1111/j.1365-2516.2004.01010.x
15. Ruiz-Sáez A. Comprehensive care in haemophilia. Hematology. 2012;17(sup1):S141–S143. doi:10.1179/102453312X13336169156492
16. Bolton-Maggs PHB. Optimal haemophilia care versus the reality. Br J Haematol. 2005;132(6):671–682. doi:10.1111/j.1365-2141.2005.05952.x
17. Soucie M, Nuss R, Evatt B, et al. Mortality among males with hemophilia: relations with source of medical care. Blood. 2000;96(2):437–442.
18. Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19(1):e1–47. doi:10.1111/j.1365-2516.2012.02909.x
19. Coppola A, Santoro C, Caterine F, et al. Emerging issues on comprehensive hemophilia care: preventing, identifying, and monitoring age-related comorbidities. Semin Thromb Hemost. 2013;39(7):794–802. doi:10.1055/s-0033-1354424
20. Yeung CHT, Santesso N, Pai M, et al. Care models in the management of haemophilia: a systematic review. Haemophilia. 2016;Suppl(3):31–40. doi:10.1111/hae.13000
21. Page D. Comprehensive care for hemophilia and other inherited bleeding disorders. Transfus Apher Sci. 2019;58(5):565–568. doi:10.1016/j.transci.2019.08.005
22. Valentino LA, Baker JR, Butler R, et al. Integrated hemophilia patient care via a national network of care centers in the United States: a model for rare coagulation disorders. J Blood Med. 2021;12:897–911. doi:10.2147/JBM.S325031
23. Astermark J, Blatný J, Königs C, Hermans C, Jiménez-Yuste V, Hart DP. Considerations for shared decision management in previously untreated patients with hemophilia A or B. Ther Adv Hematol. 2023;14:20406207231165857. doi:10.1177/20406207231165857
24. Melchiorre D, Manetti M, Matucci-Cerinic M. Pathophysiology of hemophilic arthropathy. J Clin Med. 2017;6(7):63. doi:10.3390/jcm6070063
25. Gualtierotti R, Solimeno LP, Peyvandi F. Hemophilic arthropathy: current knowledge and future perspectives. J Thromb Haemost. 2021;19(9):2112–2121. doi:10.1111/jth.15444
26. Zhao L, Yanf H, Li Y, et al. Joint status and related risk factors in patients with severe hemophilia A: a single-center cross-sectional study. Hematology. 2022;27(1):80–87. doi:10.1080/16078454.2021.2019892
27. Blanchette VS, Key NS, Ljung LR, et al. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12(11):1935–1939. doi:10.1111/jth.12672
28. Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26(Suppl. 6):1–158. doi:10.1111/hae.14046
29. Stimec J, Dover S, Pullenayegum E, et al. Magnetic resonance imaging in boys with severe hemophilia A: serial and end-of-study findings from the Canadian Hemophilia Primary Prophylaxis Study. Res Pract Thromb Haemost. 2021;5(7):e12565. doi:10.1002/rth2.12565
30. Mancuso ME, Holstein K, O’Donnell JS, Lobet S, Klamroth R. Synovitis and joint health in patients with haemophilia: statements from a European e-Delphi consensus study. Haemophilia. 2023;29(2):619–628. doi:10.1111/hae.14734
31. Jackson MD, O’Brien SH, Stanek J, Dunn AL, Kerlin BA. Inpatient health care utilization in children with hemophilia before and after the Joint Outcome Study publication. J Pediatr Hematol Oncol. 2019;41(5):e284–289. doi:10.1097/MPH.0000000000001329
32. Warren BB, Thornhill D, Stein J, et al. Young adult outcomes of childhood prophylaxis for severe hemophilia A: results of the Joint Outcome Continuation Study. Blood Adv. 2020;4(11):2451–2459. doi:10.1182/bloodadvances.2019001311
33. van Leeuwen FHP, Timmer MA, de Jong PA, Fischer K, Foppen W. Screening for subclinical synovial proliferation in haemophilia: a systematic review and meta-analysis comparing physical examination and ultrasound. Haemophilia. 2023;29(2):445–455. doi:10.1111/hae.14737
34. Gooding R, Thachil J, Alamelu J, Motwani J, Chowdary P. Asymptomatic joint bleeding and joint health in hemophilia: a review of variables, methods, and biomarkers. J Blood Med. 2021;12:209–220. doi:10.2147/JBM.S304597
35. Martinoli C, Alberighi ODC, Di Mino G, et al. Development and definition of a simplified scanning procedure and scoring method for Haemophilia Early Arthropathy Detection with Ultrasound (HEAD-US). Thromb Haemost. 2013;109(6):1170–1179. doi:10.1160/TH12-11-0874
36. Prasetyo M, Moniqa R, Tulaar A, Prihatono J, Setiawan SI. Correlation between hemophilia early arthropathy detection with ultrasound (HEAD-US) score and hemophilia joint health score (HJHS) in patients with hemophilic arthropathy. PLoS One. 2021;16(4):e0248952. doi:10.1371/journal.pone.0248952
37. von Drygalski A, Moore RE, Nguyen S, et al. Advanced hemophilic arthropathy: sensitivity of soft tissue discrimination with musculoskeletal ultrasound. J Ultrasound Med. 2018;37(8):1945–1956. doi:10.1002/jum.14541
38. Volland LM, Zhou JY, Barners RFW, et al. Development and reliability of the joint tissue activity and damage examination for quantitation of structural abnormalities by musculoskeletal ultrasound in hemophilic joints. J Ultrasound Med. 2019;38(6):1569–1581. doi:10.1002/jum.14846
39. Nacca CR, Harris AP, Tuttle JR. Hemophilic arthropathy. Orthopedics. 2017;40:e940–e946. doi:10.3928/01477447-20170619-05
40. Peyvandi F, Garagiola I, Young G. The past and future of haemophilia: diagnosis, treatments, and its complications. Lancet. 2016;388(10040):187–197. doi:10.1016/S0140-6736(15)01123-X
41. Heijnen L. Rehabilitation and the role of the physical therapist. Southeast Asian J Trop Med Public Health. 1993;24(Suppl. 1):26–29.
42. Lobet S, Hermans C, Stephensen D. The emerging clinical and scientific role of the physiotherapist in haemophilia care. Haemophilia. 2020;26(4):560–562. doi:10.1111/hae.14096
43. Boccalandro EA, Begnozzi V, Garofalo S, Pasca S, Peyvandi F. The evolution of physiotherapy in the multidisciplinary management of persons with haemophilia (PWH): a scoping review. Haemophilia. 2023;29(1):11–20. doi:10.1111/hae.14661
44. Newman JR, Durben N, Baumann K, et al. Physical therapy within US HTCs: a multicentre survey of utilization, practice patterns and pain management approaches. Haemophilia. 2022;28(2):343–350. doi:10.1111/hae.14501
45. Chimenti RL, Frey-Law LA, Sluka KA. A mechanism-based approach to physical therapist management of pain. Phys Ther. 2018;98(5):302–314. doi:10.1093/ptj/pzy030
46. Colvin BT, Astermark J, Fischer K, et al. European principles of haemophilia care. Haemophilia. 2008;14(2):361–374. doi:10.1111/j.1365-2516.2007.01625.x
47. De la Corte-Rodriguez H, Rodriguez-Merchan EC, Alvarez-Roman MT, Jiménez-Yuste V. Applying World Health Organization 2020 guidelines on physical activity and sedentary behavior to people with hemophilia. Expert Rev Hematol. 2021;14(5):429–436. doi:10.1080/17474086.2021.1924054
48. Mulder K, McCabe E, Strike K, Nilson J. Developing clinical practice guidelines for physiotherapists working with people with inherited bleeding disorders. Haemophilia. 2021;27(4):674–682. doi:10.1111/hae.14327
49. Mannucci PM. Hemophilia therapy: the future has begun. Haematologica. 2020;105(3):545–553. doi:10.3324/haematol.2019.232132
50. Nathwani AC. Gene therapy for hemophilia. Hematology Am Soc Hematol Educ Program. 2019;2019(1):1–8. doi:10.1182/hematology.2019000007
51. Leebeek FWG, Miesbach W. Gene therapy for hemophilia: a review on clinical benefit, limitations, and remaining issues. Blood. 2021;138(11):923–931. doi:10.1182/blood.2019003777
52. Stephensen D, de Kleijin P, Matlary RED, et al. Scope of practice of haemophilia physiotherapists: a European survey. Haemophilia. 2019;25(3):514–520. doi:10.1111/hae.13727
53. Parker EJ, Jamieson LM, Broughton J, Albino J, Lawrence HP, Roberts-Thomson K. The oral health of indigenous children: a review of four nations. J Paediatr Child Health. 2010;46(9):483–486. doi:10.1111/j.1440-1754.2010.01847.x
54. Ship JA, Duffy V, Jones JA, Langmore S. Geriatric oral health and its impact on eating. J Am Geriatr Soc. 1996;44(4):456–464. doi:10.1111/j.1532-5415.1996.tb06419.x
55. Zaliuniene R, Peciuliene V, Brukiene V, Aleksejuniene J. Hemophilia and oral health. Stomatologija. 2014;16(4):127–131.
56. Kanjani V, Annigeri RG, Hanagavadi S, Manjunath MR. Comparative analysis of oral health and treatment necessities in hemophilia individuals of Davangere population - a case control study. J Family Med Prim Care. 2020;9(9):4774–4777. doi:10.4103/jfmpc.jfmpc_413_20
57. Brecher EA, Lewis CW. Infant oral health. Pediatr Clin North Am. 2018;65(5):909–921. doi:10.1016/j.pcl.2018.05.016
58. Czajkowska S, Rupa-Matysek J, Gil L, Surdacka A. Assessment of oral health and healthy habits in adult patients with congenital hemophilia. Eur J Dent. 2023;17(1):161–172. doi:10.1055/s-0042-1743156
59. Baskirt EA, Zulfikar GAk G, Zulfikar B. Oral and general health-related quality of life among young patients with haemophilia. Haemophilia. 2009;15(1):193–198. doi:10.1111/j.1365-2516.2008.01919.x
60. Anderson JA, Brewer A, Creagh D, et al. Guidance on the dental management of patients with haemophilia and congenital bleeding disorders. Br Dent J. 2013;215(10):497–504. doi:10.1038/sj.bdj.2013.1097
61. Chaichareon P, Im-Erbsin T. Comprehensive care of hemophilia: role of the dentist. Southeast Asian J Trop Med Public Health. 1993;24(Suppl.1):34–36.
62. Masthoff M, Gerwing M, Masthoff M, et al. Dental imaging - a basic guide for the radiologist. Rofo. 2019;191(3):192–198. doi:10.1055/a-0636-4129
63. Karki C. Study of young people attending an adolescent friendly centre. Kathmandu Univ Med J. 2004;2(4):324–330.
64. Breakey VR, Blanchette VS, Bolton-Maggs PHB. Towards comprehensive care in transition for young people with haemophilia. Haemophilia. 2010;16(6):848–857. doi:10.1111/j.1365-2516.2010.02249.x
65. Blum RW, Garell D, Hodgman CH, et al. Transition from child-centered to adult health-care systems for adolescents with chronic conditions. A position paper of the society for adolescent medicine. J Adolesc Health. 1993;14(7):570–576. doi:10.1016/1054-139x(93)90143-d
66. Quon D, Reding M, Guelcher C, et al. Unmet needs in the transition to adulthood: 18- to 30-year-old people with hemophilia. Am J Hematol. 2015;90(Suppl2):S17–S22. doi:10.1002/ajh.24219
67. Sun J, Zhou X, Hu N. Factor VIII replacement prophylaxis in patients with hemophilia A transitioning to adults: a systematic literature review. Orphanet J Rare Dis. 2021;16(1):287. doi:10.1186/s13023-021-01919-w
68. van Galen KPM, d’Oiron R, James P, et al. A new hemophilia carrier nomenclature to define hemophilia in women and girls: communication from the SSC of the ISTH. J Thromb Haemost. 2021;19(8):1883–1887. doi:10.1182/bloodadvances.2021006388
69. Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AHJT, et al. Bleeding in carriers of hemophilia. Blood. 2006;108(1):52–56. doi:10.1182/blood-2005-09-3879
70. Fijnvandraat K, Cnossen MH, Leebeek FWG, Peters M. Diagnosis and management of haemophilia. BMJ. 2012;344:e2707. doi:10.1136/bmj.e2707
71. Lee CA, Chi C, Pavord SR, et al. The obstetric and gynaecological management of women with inherited bleeding disorders--review with guidelines produced by a taskforce of UK haemophilia centre doctors’ organization. Haemophilia. 2006;12(4):301–336. doi:10.1111/j.1365-2516.2006.01314.x
72. Inaba H, Shinozawa K, Amano K, Fukutake K. Identification of deep intronic individual variants in patients with hemophilia A by next-generation sequencing of the whole factor VIII gene. Res Pract Thromb Haemost. 2017;1(2):264–274. doi:10.1002/rth2.12031
73. Dardik R, Janczar S, Lalezari S, et al. Four decades of carrier detection and prenatal diagnosis in hemophilia A: historical overview, state of the art and future directions. Int J Mol Sci. 2023;24(14):11846. doi:10.3390/ijms241411846
74. Coppola A, Cerbone AM, Mancuso G, Mansueto MF, Mazzini C, Zanon E. Confronting the psychological burden of haemophilia. Haemophilia. 2011;17(1):21–27. doi:10.1111/j.1365-2516.2010.02280.x
75. Broderick CR, Herbeart RD, Latimer J, et al. Association between physical activity and risk of bleeding in children with hemophilia. JAMA. 2012;308(14):1452–1459. doi:10.1001/jama.2012.12727
76. van Balen M, O’Mahony B, Cnossen MH, et al. Patient-relevant health outcomes for hemophilia care: development of an international standard outcomes set. Res Pract Thromb Haemost. 2021;5(4):e12488. doi:10.1002/rth2.12488
77. Kempton CL, Stout MM, Barry V, et al. Validation of a new instrument to measure disease-related distress among patients with haemophilia. Haemophilia. 2021;27(1):60–68. doi:10.1111/hae.14187
78. Hermans C, Noone D, Benson G, et al. Hemophilia treatment in 2021: choosing the”optimal” treatment using an integrative, patient-oriented approach to shared decision-making between patients and clinicians. Blood Rev. 2022;52:100890. doi:10.1016/j.blre.2021.100890
79. Page D. Setting the scene: historical overview of challenges and what led to advances in comprehensive care in developed countries, the Canadian experience. Haemophilia. 2020;26(Suppl 3):4–5. doi:10.1111/hae.13885
80. Lee D, Le AO, Meganck M, Chamberland S, Pai A. Adding a clinical hemophilia pharmacist to the hemophilia comprehensive care model improves health care-related outcomes and drug-related costs in an integrated health care system. Perm J. 2022;26(3):90–93. doi:10.7812/TPP/21.192
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.