")
Back to Journals » Biologics: Targets and Therapy » Volume 16
Emerging Therapies for Huntington’s Disease – Focus on N-Terminal Huntingtin and Huntingtin Exon 1
Authors van der Bent ML, Evers MM, Vallès A
Received 10 June 2022
Accepted for publication 14 September 2022
Published 30 September 2022 Volume 2022:16 Pages 141—160
DOI https://doi.org/10.2147/BTT.S270657
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shein-Chung Chow
M Leontien van der Bent, Melvin M Evers, Astrid Vallès
uniQure biopharma B.V., Department of Research and Development, Amsterdam, the Netherlands
Correspondence: Astrid Vallès, uniQure biopharma B.V, Postbus 22506, Amsterdam, 1100 DA, the Netherlands, Tel +31 20 240 6000, Email [email protected]
Abstract: Huntington’s disease is a devastating heritable neurodegenerative disorder that is caused by the presence of a trinucleotide CAG repeat expansion in the Huntingtin gene, leading to a polyglutamine tract in the protein. Various mechanisms lead to the production of N-terminal Huntingtin protein fragments, which are reportedly more toxic than the full-length protein. In this review, we summarize the current knowledge on the production and toxicity of N-terminal Huntingtin protein fragments. Further, we expand on various therapeutic strategies targeting N-terminal Huntingtin on the protein, RNA and DNA level. Finally, we compare the therapeutic approaches that are clinically most advanced, including those that do not target N-terminal Huntingtin, discussing differences in mode of action and translational applicability.
Keywords: Huntingtin, N-terminal fragments, proteolysis, aberrant splicing, exon1 fragment, Huntington’s disease therapeutics
Introduction
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder with an estimated prevalence of up to 9 per 100,000 in the USA, Canada, Oceania, and Western Europe.1,2 HD is caused by a CAG (cytosine, adenine, and guanine) repeat expansion in exon 1 of the Huntingtin (HTT) gene, resulting in the translation of a mutant Huntingtin protein harboring a toxic polyglutamine (polyQ) stretch at its amino (N) terminus. Gene carriers with repeats between 36 and 39 CAG show incomplete penetrance, while repeats of 40 and more triplets lead to fully penetrant disease. The age of onset is inversely correlated with the CAG repeat length, with an average age of onset of 35–44 years. HD is characterized by motor, cognitive and psychiatric symptoms and is ultimately fatal, with a median survival of 15–18 years after onset. About 5–10% of HD patients show disease onset before 20 years of age, in which case it is called juvenile HD. Juvenile HD has a different clinical presentation compared to adult onset HD, characterized by symptoms such as severe mental retardation, speech and language delay, as well as more pronounced motor and cerebellar symptoms and overall more rapid disease progression.3
Apart from the inherited CAG length, several genetic modifiers have been identified that are associated with age of onset. Many of these modifiers point towards an important role for somatic instability: the process in which the CAG repeat within cells expands over time. Within the HTT locus, a strong genetic modifier is whether or not a CAA (cytosine, adenine, and adenine) interruption is present at the 3’ end of the CAG repeat. Similar to CAG triplets, CAA encodes for glutamine, thus resulting in the same polyQ stretch. Nonetheless, alleles that lack this CAA interruption were found to be more prone to somatic expansion and showed decreased age of onset, while the presence of an additional CAA interruption was found to delay both somatic expansion and age of onset.4,5 Moreover, many of the identified trans-acting genetic modifiers, such as FANCD2 And FANCI Associated Nuclease 1 (FAN1) and MutL Homolog 1 (MLH1), are involved in DNA mismatch repair and influence somatic instability of the CAG repeat.5,6
Although HD was initially thought to be mainly a protein toxic gain-of-function disorder, it is likely that protein loss-of-function also plays a role, as reviewed elsewhere,7–10 and there is increasing evidence for the involvement of other disease mechanisms, such as repeat-associated non-AUG dependent (RAN) translation and RNA toxic gain-of-function, also reviewed previously.11–13 Still, little is known regarding the relative contribution of each of these pathogenic mechanisms to the disease (Figure 1).
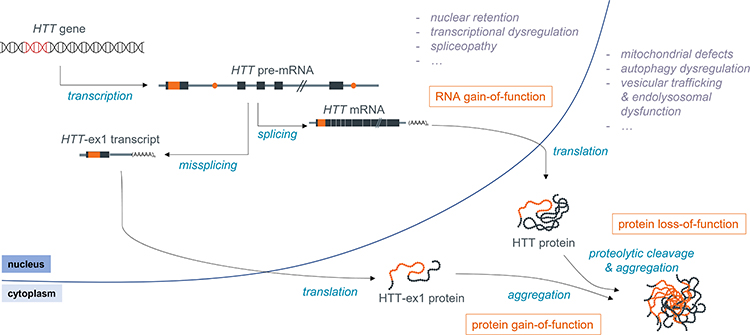 |
Figure 1 Schematic overview of the molecular pathogenesis of HD. |
HTT is known to be essential for embryonic development, as demonstrated by the fact that knockout mice are embryonically lethal, and also appears to play a role in later stages of development and life, as reviewed by Kaemmerer and Grondin.10 There is, however, no clear consensus on the level of wild type HTT (wtHTT) that is required for its normal function, as this is likely to depend on many factors, including age and tissue/brain region. wtHTT is involved in many important cellular processes, including endocytosis and vesicular trafficking, cell division, autophagy and transcriptional regulation (reviewed by Saudou and Humbert)9 which may all be impacted by a loss of wtHTT function in HD.
Compelling evidence for the involvement of RNA-mediated toxicity was provided by Sun et al, who found that even in the absence of translation, there was still repeat-length dependent toxicity of 5’ HTT mRNA as well as full-length HTT.14 RNA toxic gain-of-function is caused by the interaction between RNA-binding proteins (RBPs), such as Muscleblind like splicing regulator 1 (MBNL1) and Pre-mRNA processing factor 8 (PRPF8), and the secondary structure formed by the expanded CAG repeat in the mRNA, affecting the splicing of a range of transcripts.15,16 This interaction appears to be dependent on the purity of the CAG repeat (ie, the absence of CAA interruptions), as Mbnl1 was found to be recruited to nuclear foci in the novel BAC-CAG mouse model, which has an uninterrupted repeat, but not in the BACHD model, which harbors an interrupted repeat.17
Finally, the presence of the expanded CAG repeat has also been shown to induce repeat-associated non-AUG dependent (RAN) translation, which leads to the production of homopolymers other than polyQ that may also negatively impact cell function. RAN translation products have been detected in the affected brain regions of patients, as well as in N171-82Q mice and a C. elegans model.18,19 However, the actual contribution of RAN translation products to HD is not clear, as, for example, no RAN toxicity was observed in HD140Q knock-in mice.20
The expanded polyQ-containing mutant HTT (mHTT) protein has been shown to interact aberrantly with a variety of proteins, including transcriptional regulators such as RNA polymerase II subunit A (POLR2A), Tumor protein p53, Mouse double minute 2 (MDM2), CREB-binding protein (CBP) and Heat shock protein 70 (HSP70), cell cycle regulators like Ras homolog enriched in brain (Rheb) and mammalian target of rapamycin (mTOR), and cytoskeleton proteins such as actin and neurofilament light (NF-L). These aberrant interactions result in a complex and widespread molecular pathology, affecting many essential processes in the cell, including DNA damage repair, transcriptional regulation, mitochondrial function and apoptosis.21–25 Importantly, premature polyadenylation of the pre-mRNA as well as proteolytic cleavage of HTT protein lead to the production of a variety of HTT fragments, and there is ample evidence that such fragments, especially the short N-terminal species, are more toxic than the full-length mHTT protein.26–35 In order to make tailored therapeutics towards the short toxic fragments, a good understanding of the mechanisms leading to their formation is needed. In this review, we therefore focus on how toxic N-terminal HTT protein species are produced and how they are linked to toxicity, as well as on therapeutic strategies that are capable of reducing these fragments.
Production of Toxic N-Terminal HTT Protein Species
N-terminal HTT protein fragments are mainly produced through two distinct processes: proteolytic cleavage and premature polyadenylation (see Figure 2 and Table 1).
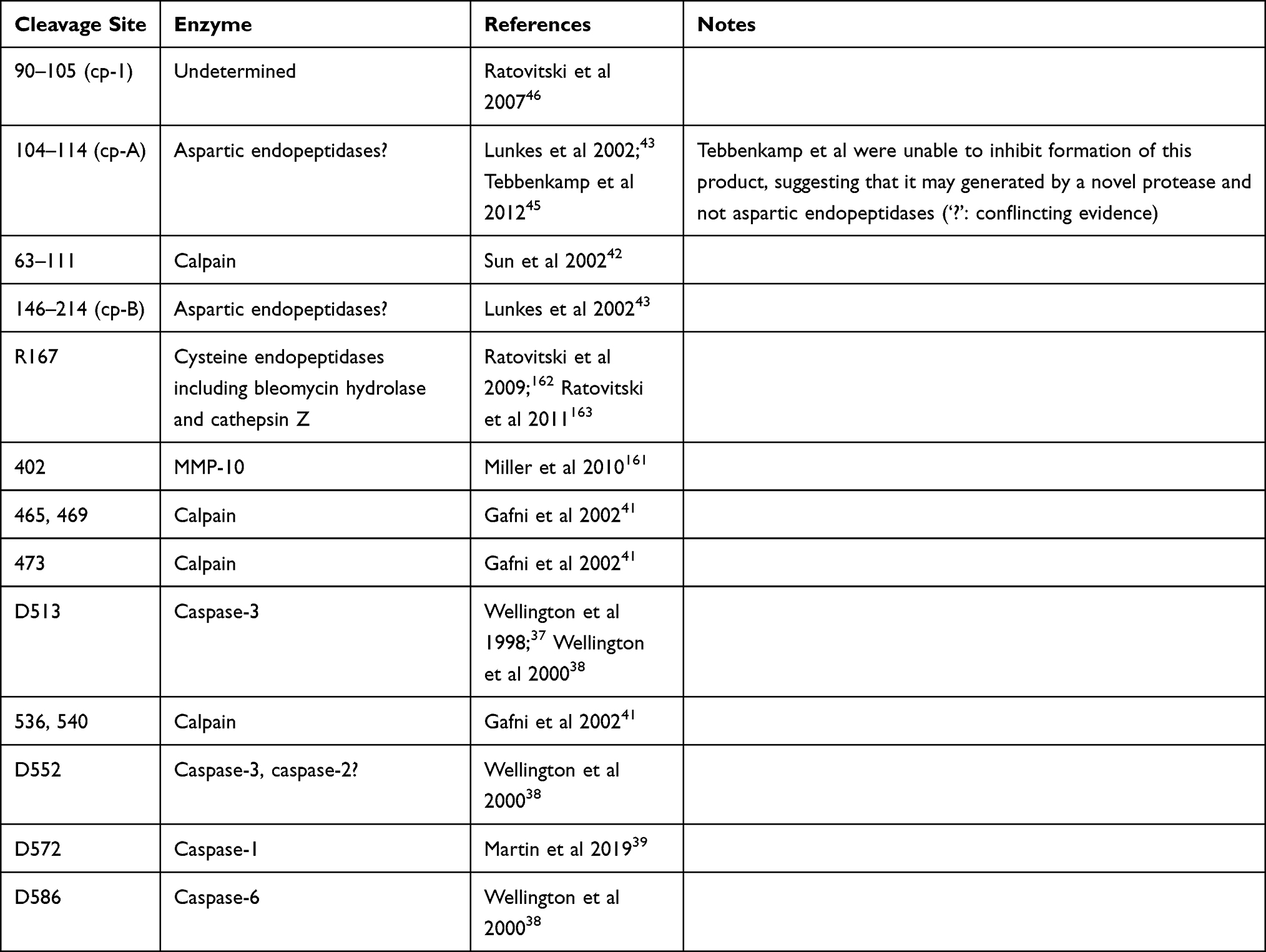 |
Table 1 Overview of Proteolytic Cleavage Sites |
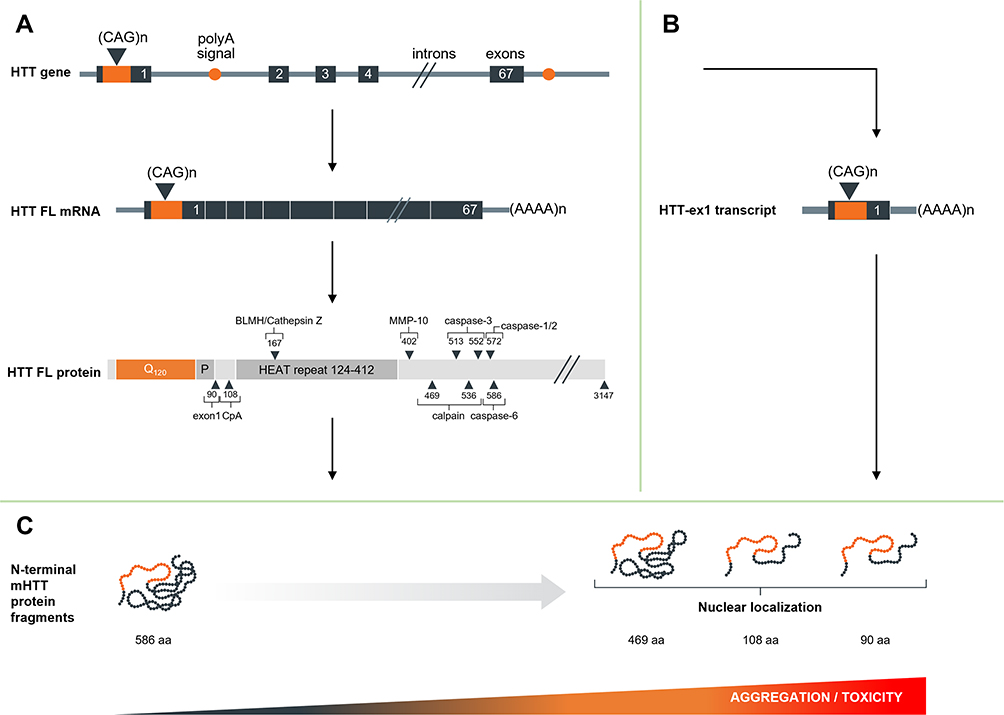 |
Figure 2 Schematic overview of production of N-terminal HTT protein. (A) Regular splicing, overview of the resulting mRNA and full-length protein and the identified proteolytic cleavage sites. (B) Alternative splicing and premature polyadenylation and resulting transcript. (C) Resulting protein species and propensity for nuclear entry, aggregation and toxicity. |
Proteolytic Cleavage
Caspase Cleavage
The group of Michael Hayden first showed that HTT could be cleaved proteolytically by apopain (caspase-3) in a repeat-length dependent manner.36 This was confirmed in a follow-up study, in which they mapped one of the caspase-3 cleavage sites to D513 and another site C-terminally of amino acid (aa) 548. Furthermore, two caspase-1 cleavage sites were identified in the first 548 aa. In contrast to their previous work with truncated HTT, the authors found no repeat-length dependence of cleavage efficiency of full-length HTT.37 In a third study, the authors were able to map the second caspase-3 cleavage site to D552, and further identified a caspase-6 cleavage site at D586.38 More recently, Martin et al recently identified yet another caspase cleavage site at D572, which was shown to be cleaved by caspase-1 and caspase-2.39
Calpain Cleavage
Both full-length and N-terminal caspase-cleavage products of HTT were found to be substrates for cleavage by calpains.40–42 Four calpain cleavage sites have been mapped, at aa 437, 465/469 and 536/54041 and between aa 63–111,42 calpain cleavage efficiency appears to be positively correlated with repeat length.41,42 Furthermore, it was shown that calpain levels, and in particular the active form, were increased in the caudate of HD patients compared to controls.41
Other Proteases/Unidentified Mechanisms
Next to caspase and calpain generated fragments, various other cleaved HTT products have been described. Lunkes et al identified two N-terminal HTT fragments, cp-A and cp-B, which appeared to be generated in transfected NG108 cells through cleavage by aspartic endopeptidases. The C-terminus of HTT cp-A fragment was mapped between aa 104–114. N-terminal fragments with the same immunogenic properties were identified in nuclear inclusions in post mortem frontal cortex of HD patients.43
Similarly, Schilling et al identified an N-terminal fragment ending between aa 90–115 in post mortem tissues from HD patients and N171-82Q mice, as well as in transfected HEK293 cells.44 Further investigation in a HEK293 cell model revealed that short, HTT cp-B-like fragments were efficiently processed to HTT cp-A-like fragments, while longer HTT fragments proved to be inefficient substrates. The C-terminus of the HTT cp-A-like fragments was mapped between aa 105 and 115–124. Although similar in size to the fragment described by Lunkes et al, inhibition of aspartyl proteases did not affect the formation of the cp-A-like fragment, and the authors were unable to identify any protease that generates these HTT cp-A-like fragments, suggesting that i) the fragments are not the same or ii) that the cp-A-like fragment described by Schilling et al is the same fragment but generated by a novel protease, which may be cell-type dependent.45 Ratovitski et al identified two N-terminal fragments (HTT cp-1 and cp-2) in PC12 and HEK293 cells expressing full-length HTT with 21Q or 126–153Q or a truncated N1212 HTT fragment with 15Q or 138Q.46 These fragments were similar in size to the previously described HTT cp-A and cp-B fragments but were not affected by inhibition of aspartic endopeptidases. In addition, they were not affected by deletion of aa 105–114. In combination with the epitope mapping, this narrowed the C-terminus of the HTT cp-1 fragment down to between aa 90 and 105, shorter than the cp-A and cp-A-like fragments described by Lunkes et al43 and Schilling et al44,45 Based on the absence of identified proteases and on the fragment length, we speculate that the generation of these fragments could involve aberrant splicing (see Aberrant Splicing and Premature Polyadenylation), although this would require further investigation.
Finally, Landles et al showed fourteen different N-terminal HTT protein isoforms (fragments 1–14) in brain tissue from HdhQ150 KI mice, the three shortest of which (fragments 12–14) were specific to mHTT.33 Some of these fragments could be linked to specific proteolytic cleavage events: fragment 7 terminated at a novel calpain cleavage site between aa 510–654, fragment 8 appeared to correspond to the D586 caspase-6 cleavage product, fragment 9 was likely produced by cleavage at calpain site 536 and fragment 10 by caspase cleavage at D513. Lastly, fragment 13 was determined to correspond to HTT-ex1.
In summary, many different proteases have been found to act on mHTT and wtHTT, generating N-terminal and C-terminal HTT fragments. The availability of antibodies that can recognize these fragments, as well as the possibility to specifically inhibit certain proteases, have allowed mapping of various fragments, albeit with variable resolution. Nonetheless, for multiple fragments, the mechanisms of production remain to be identified.
Aberrant Splicing and Premature Polyadenylation
Besides proteolytic cleavage, there are other mechanisms that lead to the generation of toxic N-terminal mHTT fragments. Sathasivam et al showed that incomplete splicing of intron 1 leads to the production of a short premature polyadenylated HTT-ex1 transcript in various HD mouse models and that this HTT-ex1 can be translated into a 90 aa N-terminal HTT-ex1 protein (based on 23Q). HTT-ex1 transcript was also found to be expressed in HD patient fibroblasts and cortex.47 In a follow-up study, Neueder et al confirmed that the HTT-ex1 transcript can be detected in patient-derived fibroblasts, as well as HD patient cerebellum, sensory motor cortex and hippocampus, with the highest expression levels measured in juvenile HD patient tissues.48 The HTT-ex1 transcript has also been detected by RNA-sequencing in various HD mouse models, including BACHD, BAC-CAG and HdhQ111.17 Both in vitro and in patient-derived tissues, the production of the HTT-ex1 transcript appears to be positively correlated with CAG repeat length, showing much higher expression in cells and tissues derived from juvenile HD patients.48,49
The current hypothesis is that HTT-ex1 formation is influenced by a combination of sequestration of spliceosome components such as U1 snRNP at the CAG repeat, leading to less efficient splicing of exon 1 to exon 2, and a reduced transcription rate, which leads to longer exposure of the cryptic polyA site in intron 1. Although the Bates group initially found evidence for the involvement of Serine and Arginine Rich Splicing Factor 6 (SRSF6) in HTT-ex1 formation,47,49 they later found that the silencing of Srsf6 in HD mouse models did not affect HTT-ex1 formation.50 It has therefore been hypothesized that multiple RNA-binding proteins may be involved in the missplicing of HTT-ex1.12 Regardless of the exact mechanisms involved, aberrant mHTT splicing is CAG repeat length dependent, suggesting that HTT-ex1 formation and associated toxicity would increase as somatic instability progresses in HD48 and that interventions targeting repeat expansion and HTT-ex1 may have therapeutic advantage.
Properties of N-Terminal Protein Species
Consistently accumulating evidence indicates that small N-terminal fragments containing extended polyQ tracts significantly contribute mHTT cellular mislocalization, aggregation and toxicity. Initial studies by the Ross group showed that transfection of N2a or HEK293 cells with full-length HTT with either 23Q or 82Q, or of truncated HTT N171-18Q or N63-18Q resulted in a diffuse cytoplasmic localization of the protein. In contrast, transfection with N171-82Q or N63-82Q led to more punctate labeling in both cytoplasm and nucleus, with the short N63-82Q construct showing the most prominent nuclear localization.51 The Hayden group found similar results, showing that N-terminal fragments of 427, 548 or more aa formed mainly perinuclear aggregates, while fragments up to 224 aa showed both cytoplasmic and nuclear aggregates. Furthermore, they found that pathogenicity depended both on repeat length and on fragment size.26,27
Barbaro et al found that, in Drosophila, shorter N-terminal fragments were more toxic and more prone to aggregate, with HTT-ex1 being by far the most toxic species.28 In mice, the R6/2 model that expresses only HTT-ex1 is by far the most swiftly progressing HD mouse model,52,53 while conditional suppression of HTT-ex1 has been shown to be neuroprotective.54 Recent in vitro studies by the Lashuel group confirm these results and further extend the findings by showing that the polyQ and Nt17 domains of HTT-ex1 synergistically modulate the aggregation propensity of HTT-ex1, with a key role of the Nt17 domain in regulating HTT-ex1 aggregation dynamics and subcellular localization and toxicity.34
There is conflicting evidence with regard to the pathogenicity of nuclear and cytoplasmic mHTT. Some groups have reported evidence that nuclear localization is required for toxicity. For example, the Greenberg group showed that adding a nuclear export signal to a N171 HTT fragment blocked its toxicity in transfected striatal neurons.55 In contrast, the Hayden group reported that neither the addition of a nuclear localization signal to a N548 HTT fragment nor the addition of a nuclear export signal to a N151 fragment altered the toxicity of those fragments, suggesting that both the nucleus and the cytoplasm are sites of HD toxicity.56 Trushina et al found that nuclear entry of mHTT only occurred after commitment of a cell to cell death. Therefore, the authors argue that nuclear mHTT localization may not be the primary event leading to toxicity.57
Intranuclear and neuropil aggregates have been observed in most HD animal models,17,30,31,58–63 and the presence of aggregates containing N-terminal HTT fragments has also been confirmed in patient brains by multiple groups.40,64,65 However, various groups have shown that it is not the insoluble aggregates or inclusion bodies, but rather the soluble oligomers that are the more toxic species.66–69 In fact, some groups have found evidence that the formation of intranuclear inclusions may be protective,55,70,71 as reviewed by Arrasate and Finkbeiner.72 Mechanistically, this may be explained by the fact that soluble mHTT-ex1 oligomers have more aberrant protein interactions than insoluble aggregates and inclusions.73 Importantly, the length of N-terminal protein species and the associated sequence context, as well as post-translational modifications, also appear to play an important role in the aggregation process.35,74,75 For more in-depth reviews on the role of post-translational modifications, we redirect elsewhere.76,77
Therapeutic Strategies to Reduce N-Terminal HTT and HTT-ex1
Various approaches have been investigated to therapeutically lower the expression or reduce the toxicity of the mutant HTT protein. The proteolytic cleavage pathway can be targeted to reduce the formation of N-terminal mHTT protein species. Furthermore, the N-terminal part of the protein can be targeted to reduce aggregation and/or increase clearance of mHTT. Finally, mHTT can be targeted at the transcript or gene level. Here, we will focus on approaches that are able to target not only full-length HTT but also HTT-ex1 and other N-terminal mHTT species, considering their potential therapeutic advantage (see Table 2).
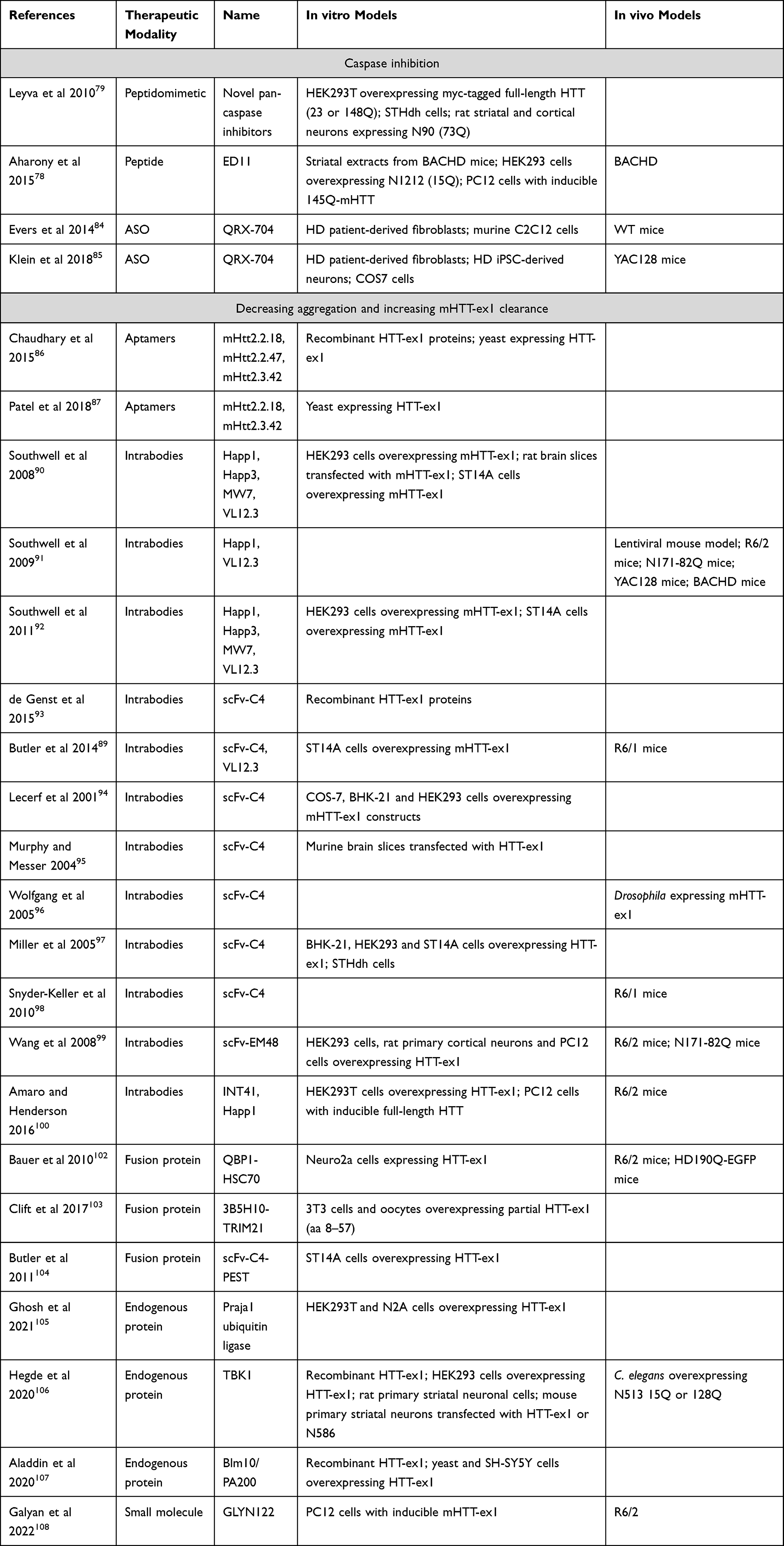 |
Table 2 Overview of Studies Targeting HTT Protein |
Reducing Proteolytic Cleavage
Caspase inhibition has been shown to reduce the proteolytic cleavage of mHTT and to improve the HD phenotype in BACHD78 and HdhQ111 mice.79 These results are backed up by earlier studies, where mutation of caspase-6 cleavage sites slowed down disease progression in YAC128 mice.80 However, it is not clear to what extent the protective effects are due specifically to the reduction of N-terminal mHTT species, rather than a general protective effect of caspase inhibition, as caspase inhibition was also protective in R6/2 and malonate models of HD, which do not express caspase-cleavable mHTT.81–83
Using a different approach, Evers et al showed that removal of the caspase-6 cleavage site by antisense oligonucleotide (ASO)-mediated skipping of (part of) exon 12 led to reduced levels of the N568 fragment in vitro and in vivo in wild type and YAC128 mice.84,85 Except for the absence of astrogliosis, no data are available regarding phenotypic effects of this ASO treatment.
None of these approaches have yet successfully been translated into the clinic, and although all may potentially decrease the formation of toxic mHTT fragments and have the potential of allele-specificity, mechanisms of RNA-associated toxicity would not be addressed.
Decreasing Aggregation and Increasing mHTT-ex1 Clearance
Aptamers
Aptamers are single-stranded oligonucleotides that, through their tertiary structure, can interact with target molecules such as proteins. The Roy lab identified aptamers that bind specifically to mHTT with 51 or 103Q but not wtHTT with 20Q.86,87 The selected aptamers were shown to inhibit aggregation of recombinant mHTT-ex1 in cell-free assays and in yeast, as well as reducing oxidative stress and mitochondrial dysfunction.86 To our knowledge, this approach has not yet been tested in vivo.
Intrabodies
Various antibodies have been expressed intracellularly as “intrabodies” to target the N-terminus of HTT. In vivo, such intrabodies are delivered using viral vectors. An excellent review on the use of intrabodies in various neurodegenerative diseases was written by Messer and Butler.88
Two groups of intrabodies have been tested most extensively (see Figure 3): those that bind to the N-terminus of HTT (VL12.3, scFv-C4) and those that recognize the proline-rich regions (PRRs) in HTT-ex1 (MW7, Happ1, Happ3, INT41). In addition, there is some literature about polyQ-binding intrabodies (MW1, MW2) and a more C-terminal intrabody derived from EM48 (scFv-EM48).
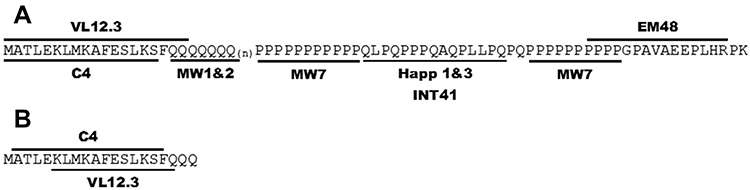 |
Figure 3 Anti-HTT Exon 1 intrabodies. (A) Antigens used to select the published anti-HTT intrabodies. (B) Specific binding identified by crystallography for scFvC4 and VL12.3. Notes: Reproduced from Messer A, Butler DC. Optimizing intracellular antibodies (intrabodies/nanobodies) to treat neurodegenerative disorders. Neurobiol Dis. 2020;134(October 2019):104619. doi: 10.1016/j.nbd.2019.104619 under Creative Commons BY-NC-ND 4.0.88 |
Southwell et al showed that intrabodies that bind to the PRR, ie, MW7, Happ1 and Happ3, increase the turnover of mHTT-ex1 overexpressed in vitro. VL12.3, an intrabody that binds to the N-terminal 17 aa of HTT, did not affect turnover, but did increase the nuclear localization of mHTT-ex1.90 In vivo, the PRR-binding Happ1 was shown to be beneficial in five different HD mouse models. In contrast, VL12.3, while effective in a lentiviral HD model, was ineffective in YAC128 mice and had a detrimental effect in R6/2 mice.91 The authors later showed that the increased turnover mediated by the PRR-binding intrabodies is dependent on a calpain-chaperone-mediated autophagy-dependent mechanism and that this process is blocked by VL12.3,92 explaining the detrimental effects of VL12.3.
Although scFv-C4 also binds to the N-terminus of HTT,93 its predominant cytoplasmic localization appears to protect from the detrimental effects observed for VL12.3.89 The scFv-C4 intrabody was shown to have beneficial effects in various HD models, including in vitro models, Drosophila and different mouse models.94–98
Two additional intrabodies have been investigated: scFv-EM48 and INT41. Like Happ1, scFv-EM48, which binds just C-terminally to the second PRR, was shown to increase turnover of mHTT, and improved motor function of N171-82Q mice.99 INT41, an intrabody that recognizes the same epitope as Happ1, but which has enhanced cytoplasmic solubility, was shown to improve cognitive function in female R6/2 mice.100
Engineered Molecules and Endogenous Proteins
In addition to the increased turnover induced by some of the intrabodies, the endogenous cellular machinery can be harnessed specifically to target proteins for degradation, using engineered proteins, peptides or small molecules. These can direct the protein of interest to the ubiquitin proteasome system, the autophagy-lysosomal pathway or chaperone-mediated autophagy. These approaches and their specific application in the context of HD have been extensively reviewed by Jarosińska and Rüdiger.101
Two such approaches specifically target the polyQ region. Bauer et al engineered a fusion molecule consisting of two copies of a polyQ-binding peptide (QBP1) and heat shock-cognate protein 70 (HSC70)-binding motifs to induce chaperone-mediated autophagy.102 Clift et al co-expressed a polyQ-binding antibody (3B5H10) with TRIM21 in an approach that they call Trim-Away, to target mHTT for proteasomal degradation.103 Additionally, Butler et al produced a fusion protein consisting of the scFv-C4 intrabody and a PEST motif to enhance proteasomal degradation of HTT-ex1.104
Several endogenous proteins have been described to enhance the turnover of mHTT, including Praja1 ubiquitin ligase,105 TBK1106 and Blm10/PA200.107 Induction or overexpression of such proteins may represent a therapeutic strategy, although, so far, this notion is only supported by experiments in cellular, Drosophila, and C. elegans models. Additionally, specificity for mHTT has not been shown for any of these three proteins.
Finally, a small molecule that can bind to mHTT-ex1, called GLYN122, has been identified recently. GLYN122 was shown to reduce mHTT-ex1 aggregation in PC12 cells, as well as reducing mHTT in cortex and striatum of R6/2 mice after intraperitoneal injection.108
Targeting HTT-ex1 at the Transcript Level
Next to targeting the pathogenic protein species itself, the production of such proteins can also be inhibited by targeting the HTT mRNA. Many different approaches have been tested to this effect, including ASOs, siRNAs, shRNAs and miRNAs (Table 3). Again, we only focus on those strategies that target HTT-ex1. Broadly speaking, the HTT-ex1 mRNA targeting approaches can be divided into those that target the expanded CAG repeat, and those that target other regions of HTT-ex1. In addition, some other approaches have been described.
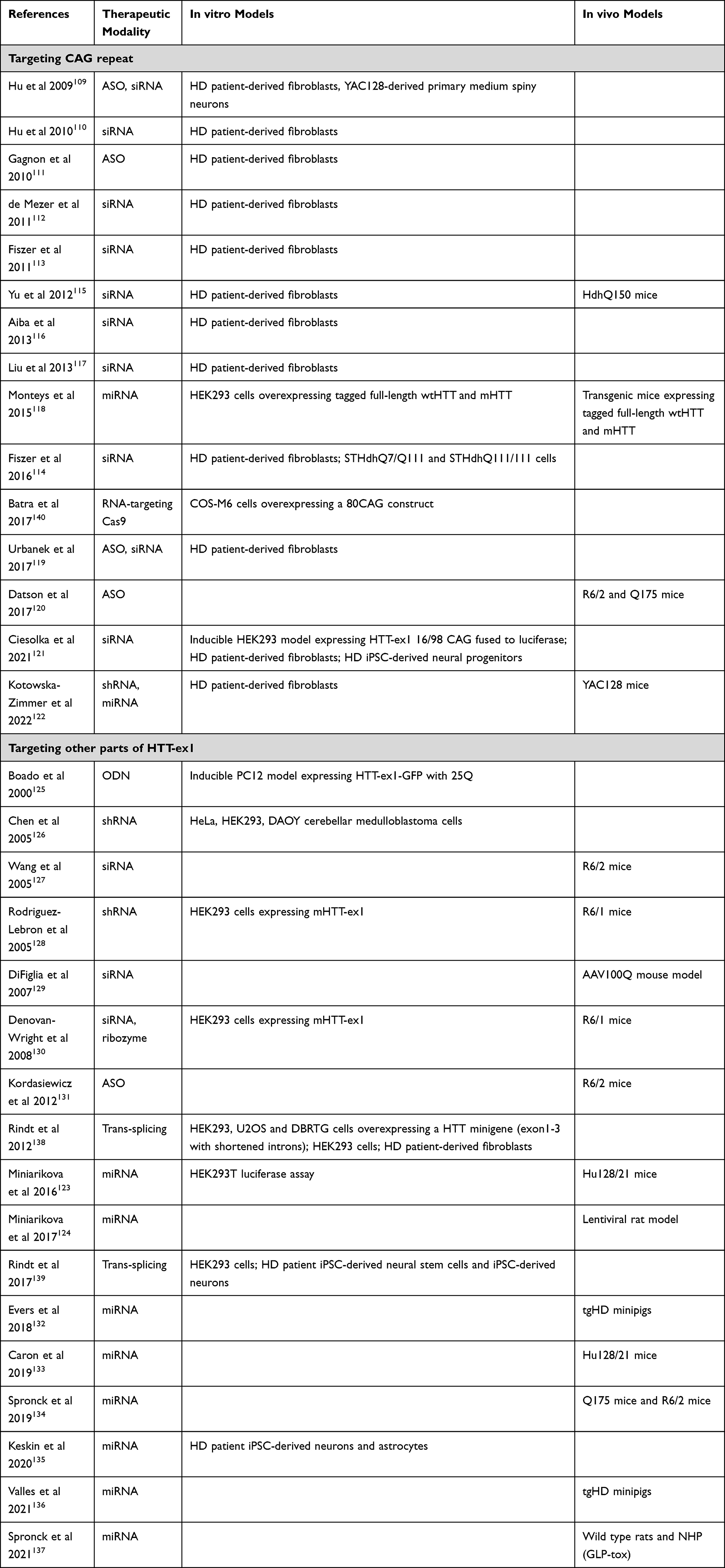 |
Table 3 Overview of Studies That Evaluated Therapeutic Approaches Targeting HTT at the RNA Level |
Antisense Oligonucleotides and RNAi Agents
Many studies have tested ASOs or RNAi agents to target the CAG repeat.109–122 In general, CAG-targeting confers preference towards the expanded allele, as this allows for binding of multiple molecules per mRNA.111 Only a few studies included in vivo efficacy. Yu et al showed the efficacy of their siRNA in HdhQ150 mice.115 Monteys et al used transgenic mice expressing tagged full-length wtHTT and mHTT, showing preferential silencing of mHTT.118 Datson et al showed the efficacy of their CAG-targeting ASO in R6/2 and Q175 mice,120 an ASO that is now further developed by Vico Therapeutics. Kotowska-Zimmer et al have shown that artificial miRNAs targeting the CAG repeat specifically reduced mHTT in YAC128 mice.122
A number of strategies that target other regions of HTT-ex1 have been described as well.123–137 This approach would be expected to lower both wtHTT and mHTT. With the exception of Boado et al and Kordasiewicz et al, who used ASOs, all of these studies utilized RNAi agents. Various groups have demonstrated efficacy of siRNA or shRNA in R6/1, R6/2 and AAV100Q mice.127–130 uniQure’s miRNA therapy has shown target engagement in the widest range of HD animal models, including Hu128/21, Q175 and R6/2 mice, lentiviral rat model and transgenic HD minipigs,123,124,132–134,136 as well as a favorable safety profile in toxicity studies in rats and non-human primates.137
Other RNA-Targeting Approaches
A handful of studies described other approaches to HTT RNA-targeting. Rindt et al developed a method to induce trans-splicing, by which mHTT exon 1 is replaced with exogenous wtHTT exon 1 in the mRNA. Thus far, there is only in vitro proof of principle for this approach, and the efficiency is rather low, with 10–15% of trans-splicing observed even after extensive optimization.138,139 Batra et al have developed an RNA-targeting Cas9 approach which targets the CAG repeat.140 For HD, there is only in vitro evidence for this approach so far, but a similar approach targeting a CUG (cytosine, uracil, and guanine) repeat was shown to be effective in vivo in myotonic dystrophy type 1 mouse models.141 This platform is being developed by Locanabio.
Finally, some small molecules have been described to bind to either HTT-ex1 or the CAG repeat, most notably furamidine, myricetin and a series of pyridocoumarin derivatives, reviewed elsewhere.12 These compounds have been described to inhibit translation of HTT. However, specificity of such compounds is generally low, thereby increasing the chance of unwanted off-target effects.
Targeting HTT-ex1 at the DNA Level
Finally, several approaches that target the HTT gene have been described (Table 4).
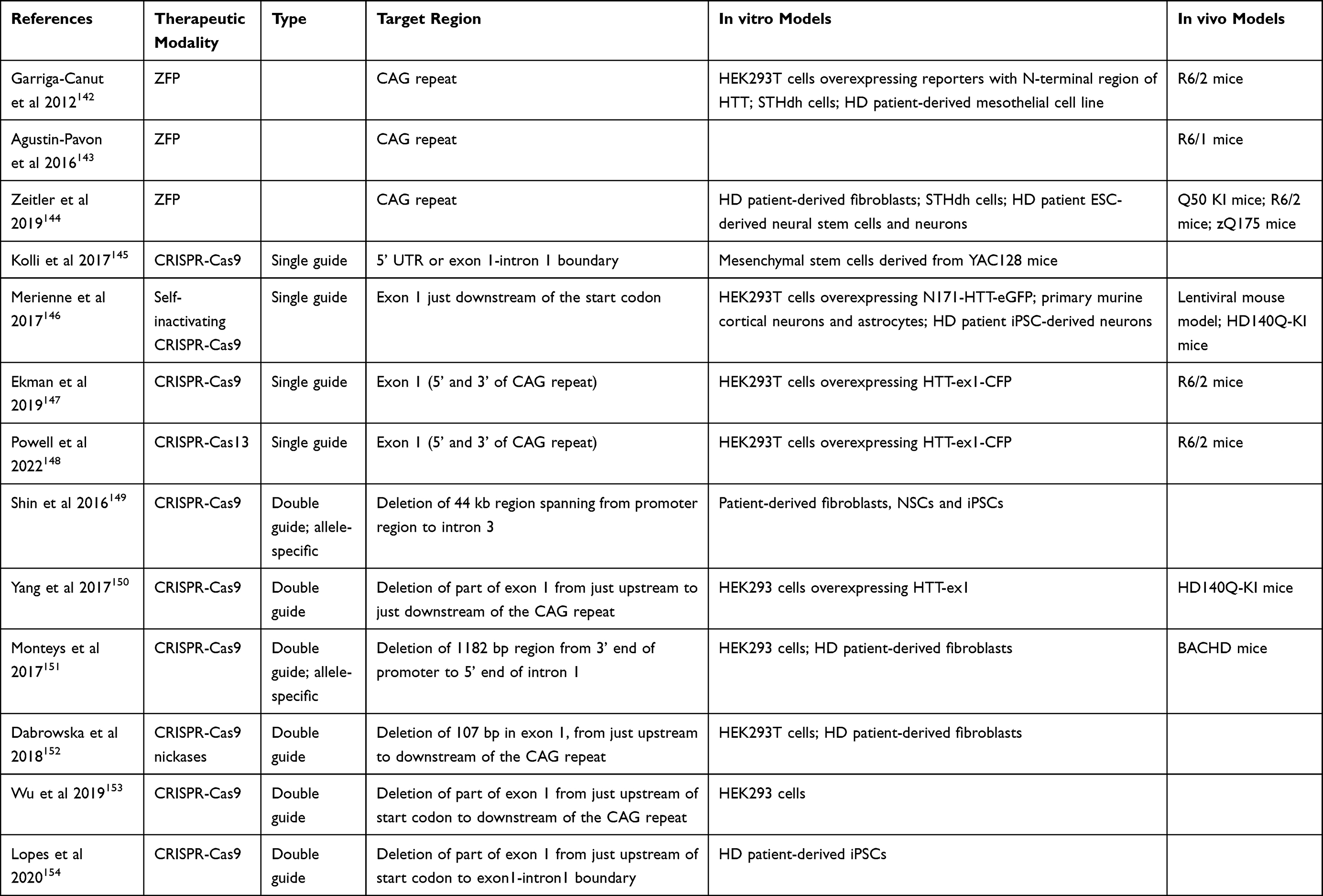 |
Table 4 Overview of Studies Targeting the HTT Gene |
Transcription can be prevented using zinc finger proteins (ZFPs) targeting the expanded CAG repeat.142–144 This approach shows allele-selectivity for the expanded repeat and is currently being developed for the clinic by Sangamo and Takeda. Further, CRISPR-Cas9 genome editing approaches have been developed to either knock out HTT by inducing mutations or excise the region containing the CAG repeat. Several groups have shown in vitro and in vivo proof of principle using single guide RNAs directed to HTT-ex1 to induce HTT knockout.145–148 Further, using a double guide RNA approach, various groups have shown that it is possible to excise the region containing CAG repeat.149–154 The size of this region differs based on the chosen guide RNAs, with the first report by Shin et al deleting a large 44 kb region,149 while the most precise excision was shown by Yang et al and Monteys et al, who deleted only the CAG repeat and small flanking regions.150,151
HTT Targeting Therapies in Clinical Development
Several HTT lowering therapies are either already in clinical trials or are close to entering the clinic. These therapies include different therapeutic modalities and mechanisms of action, each with distinct potential efficacy and safety profiles. Only the approaches in clinical trials or performing IND-enabling studies are covered here.
Two of the most advanced programs, the Phase III trial with the non-allele-specific HTT exon 36-targeting ASO tominersen (Roche) and the phase I/II trials with the allele-specific mHTT-associated single nucleotide polymorphism (SNP)-targeting ASOs WVE-120101 and WVE-120102 (Wave Life Sciences) were halted in 2021, as reviewed elsewhere.155 Roche plans to design a new Phase II study with tominersen, for younger adult patients with lower disease burden (https://ir.ionispharma.com/news-releases/news-release-details/ionis-partner-evaluate-tominersen-huntingtons-disease-new-phase). Wave Life Sciences has now initiated a new trial with their novel product WVE-003, which targets another SNP and has improved chemistry (clinicaltrials.gov NCT05032196). These ASOs are administered repeatedly through intrathecal administration, which may explain some of the adverse events observed with tominersen, which was more pronounced in the cohort receiving more frequent administration.155 Neither drug is expected to affect HTT-ex1 formation or RNA-mediated toxicity.
Novartis and PTC Therapeutics both have initiated Phase 2 clinical trials for their splicing modulators Branaplam (NCT05111249) and PTC518 (NCT05358717). These small molecules induce the inclusion of a pseudoexon between HTT exons 49 and 50, which leads to a premature stop codon and subsequent nonsense-mediated decay.156,157 One of the main advantages is that these small molecules can be administered orally. Furthermore, the mechanism of action targets the pre-mRNA and is therefore quite upstream in the molecular pathology. However, this approach is not specific for the mutant allele and, as it targets a downstream exon, is also not expected to affect HTT-ex1 production or toxic RNA gain-of-function.
In a more indirect fashion, metformin has been shown to reduce translation of HTT through interacting with the MID1/PP2A/mTOR protein complex.158 Interestingly, the effect of metformin was found to be specific for mHTT and to also impact HTT-ex1 protein formation. The drug can be administered orally, and as it is already in clinical use for the treatment of diabetes, its safety profile has already been well established. Metformin is currently being tested for the treatment of HD in a phase III clinical trial to establish its potential as a treatment for HD (NCT04826692). Although it has been shown to reduce HTT levels, RNA-mediated toxicity is not expected to be targeted by its mechanism of action.
There are no therapies that target HTT-ex1 exclusively, but some therapies target HTT-ex1 in addition to the full-length HTT. The most advanced is uniQure’s gene therapy AMT-130, which is currently being tested in phase I/II clinical trials (NCT04120493 and NCT05243017). AMT-130 is an AAV5-delivered miRNA which is administered through a one-time intrastriatal injection. This therapy is not allele-selective, and its effect on RNA-mediated toxicity has not yet been established.
Several other HTT-ex1 targeting candidates are close to entering clinical trials, including Galyan Bio’s HTT-ex1 binding small molecule GLYN122 and Vybion’s INT41 intrabody. These therapeutic candidates target the protein and are therefore not expected to impact RNA-mediated toxicity. According to the companies’ websites, both are performing IND-enabling studies, although their target date to enter the clinic is not clear (https://www.galyan.bio/pipeline, https://www.vybion.com/?page=product_pipeline).
Likewise, Vico Therapeutics received FDA orphan drug designation for their CAG-targeting ASO in July 2021 and is expected to start clinical trials soon (https://vicotx.com/pipeline/). Takeda and Sangamo are further developing their ZFP approach targeting the CAG repeat (https://www.sangamo.com/programs/). Both approaches preferentially target mHTT and as they act on the (pre-)mRNA and on transcription, respectively, these drug candidates may also have a beneficial effect on RNA-mediated toxicity.
Discussion
Although all the approaches mentioned, as well as others in earlier phases of development, aim to reduce HTT levels, their mechanism of action is different and not all pathways related to HTT toxicity will be engaged. The relative contribution of each pathway is a matter of debate and is likely to depend on many factors, including age, tissue and cell type. Several of the described mechanisms of N-terminal HTT fragment production, including calpain cleavage and premature polyadenylation, have been shown to correlate with repeat length. This is also the case with HTT-ex1 formation through aberrant splicing. Therefore, it may be expected that as the repeat gets longer over time due to somatic instability, the contribution of these mechanisms will increase. Nonetheless, the broad molecular pathology of HD would likely benefit most from an intervention that acts as far upstream as possible, ie, on the DNA or the RNA level.
For an approach to be successful in disease modification, next to efficiency, adequate safety is key. Safety issues can arise from intrinsic characteristics of the therapeutic modality itself (eg, chemistry, properties of the therapeutic vector, and need of chronic administration), which are not covered in this review. The mechanism of action of the approach can also have different safety risks. Very specific approaches, with a well-understood mechanism, and with low to no interactions with other processes and molecules other than those related to HTT toxicity, would be preferred.
Multiple different approaches are running head-to-head. The small molecule splicing modulators are among the most elegant in terms of delivery, as these are capable of crossing the blood–brain barrier and can therefore be administered orally. However, these small molecules are not specific for mHTT or even solely for HTT, and long-term studies are needed to determine the safety profile. Furthermore, these splicing modulators are expected to affect neither aberrant splicing of HTT-ex1 nor toxic RNA gain-of-function effects. ASOs and siRNAs have a less favorable distribution and need to be administered locally, although novel chemistries, such as peptide nucleic acids and di-siRNAs, have shown more promising biodistribution and may allow for systemic administration. These synthetic oligonucleotides are active for a limited amount of time, and therefore need to be readministered frequently. CAG-targeting ASOs are expected to not only reduce HTT and HTT-ex1 protein gain-of-function but also to alleviate RNA-mediated toxicity; however, non-specific effects on other genes containing CAG repeats may be difficult to overcome. Finally, the gene therapy approaches utilize AAVs to deliver their cargo. The current generation of AAVs is not sufficiently capable of crossing the blood–brain barrier and therefore still needs to be administered locally, although efforts are ongoing to identify novel capsids that could be administered in a less invasive manner, eg, Goertsen et al.159 Because most cells that are targeted in HD are non-dividing, a more invasive route of administration is, however, less of an issue, as the therapy would only need to be administered once. uniQure’s miRNA-based strategy would reduce toxic protein gain-of-function, whereas Takeda and Sangamo’s ZFP approach targets DNA and thereby acts upstream of mHTT transcription, which would improve both toxic protein- and RNA gain-of-function; yet, as the mechanism of action of this approach involves direct targeting of the repeat, off-target effects may be an issue. Pre-clinically, gene editing approaches using CRISPR-Cas are being explored. However, long-term studies will need to show the safety profiles of such approaches.
To maximize therapeutic efficacy, future research will need to point out whether it may be advantageous to combine various therapeutic strategies with different modes of action. Further, it is likely that any therapeutic approach will benefit from as early intervention as possible. To this end, excellent safety profiles and good biomarkers of both safety and efficacy will be key.160
Conclusion
In summary, we have reviewed the production of N-terminal HTT protein fragments, their role in HD pathology, as well as therapeutic approaches to target these toxic species. Extensive research into HD continues to deepen our understanding of the broad molecular mechanisms leading to disease. With the increasing understanding of the pathological mechanisms associated with mHTT, several different therapeutic approaches are being developed, which will hopefully lead, in the near future, to halting or modification of this devastating disease.
Acknowledgments
We thank our uniQure colleagues who provided a critical review of the manuscript.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
LB, ME and AV are employees of, and may own stock/options in, uniQure biopharma B.V. In addition, Dr Astrid Vallès has a patent WO2021053018 issued to UNIQURE IP B.V. The authors report no other conflicts of interest in this work.
References
1. Crowell V, Houghton R, Tomar A, Fernandes T, Squitieri F. Modeling manifest Huntington’s disease prevalence using diagnosed incidence and survival time. Neuroepidemiology. 2021;55(5):361–368. doi:10.1159/000516767
2. Rawlins MD, Wexler NS, Wexler AR, et al. The prevalence of Huntington’s disease. Neuroepidemiology. 2016;46(2):144–153. doi:10.1159/000443738
3. Caron NS, Wright GE, Hayden MR, et al. Huntington disease. In: Adam M, Ardinger H, editors. GeneReviews®. Seattle: University of Washington; 1993:1–34.
4. Wright GEB, Collins JA, Kay C, et al. Length of uninterrupted CAG, independent of polyglutamine size, results in increased somatic instability, hastening onset of Huntington disease. Am J Hum Genet. 2019;104(6):1116–1126. doi:10.1016/j.ajhg.2019.04.007
5. Lee J-M, Correia K, Loupe J; GeM-HD GM of HDC. CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell. 2019;178(4):887–900.e14. doi:10.1016/j.cell.2019.06.036
6. Iyer RR, Pluciennik A, Mismatch DNA. Repair and its role in Huntington’s disease. J Huntingtons Dis. 2021;10(1):75–94. doi:10.3233/JHD-200438
7. Schulte J, Littleton JT. The biological function of the Huntingtin protein and its relevance to Huntington’s disease pathology. Curr Trends Neurol. 2011;5:65–78.
8. Liu JP, Zeitlin SO. Is huntingtin dispensable in the adult brain? J Huntingtons Dis. 2017;6(1):1–17. doi:10.3233/JHD-170235
9. Saudou F, Humbert S. The biology of Huntingtin. Neuron. 2016;89(5):910–926. doi:10.1016/j.neuron.2016.02.003
10. Kaemmerer WF, Grondin RC. The effects of Huntingtin-lowering: what do we know so far? Degener Neurol Neuromuscul Dis. 2019;9:3–17. doi:10.2147/DNND.S163808
11. Neueder A, Bates GP. RNA related pathology in Huntington’s disease. Polyglutamine Disord. 2018;2018:85–101. doi:10.1007/978-3-319-71779-1_4
12. Heinz A, Nabariya DK, Krauss S. Huntingtin and its role in mechanisms of RNA-mediated toxicity. Toxins. 2021;13(7):487. doi:10.3390/toxins13070487
13. Malik I, Kelley CP, Wang ET, Todd PK. Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat Rev Mol Cell Biol. 2021;22(9):589–607. doi:10.1038/s41580-021-00382-6
14. Sun X, Li PP, Zhu S, et al. Nuclear retention of full-length HTT RNA is mediated by splicing factors MBNL1 and U2AF65. Sci Rep. 2015;5:1–16. doi:10.1038/srep12521
15. Mykowska A, Sobczak K, Wojciechowska M, Kozlowski P, Krzyzosiak WJ. CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Res. 2011;39(20):8938–8951. doi:10.1093/nar/gkr608
16. Schilling J, Broemer M, Atanassov I, et al. Deregulated splicing is a major mechanism of RNA-induced toxicity in Huntington’s disease. J Mol Biol. 2019;431(9):1869–1877. doi:10.1016/j.jmb.2019.01.034
17. Gu X, Richman J, Langfelder P, et al. Uninterrupted CAG repeat drives striatum-selective transcriptionopathy and nuclear pathogenesis in human Huntingtin BAC mice. Neuron. 2022;110(7):1173–1192.e7. doi:10.1016/j.neuron.2022.01.006
18. Bañez-Coronel M, Ayhan F, Tarabochia AD, et al. RAN translation in Huntington disease. Neuron. 2015;88(4):667–677. doi:10.1016/j.neuron.2015.10.038
19. Rudich P, Watkins S, Lamitina T. PolyQ-independent toxicity associated with novel translational products from CAG repeat expansions. PLoS One. 2020;15(4):1–21. doi:10.1371/journal.pone.0227464
20. Yang S, Yang H, Huang L, et al. Lack of RAN-mediated toxicity in Huntington’s disease knock-in mice. Proc Natl Acad Sci U S A. 2020;117(8):4411–4417. doi:10.1073/pnas.1919197117
21. Steffan JS, Kazantsev A, Spasic-Boskovic O, et al. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97(12):6763–6768. doi:10.1073/pnas.100110097
22. Suhr ST, Senut MC, Whitelegge JP, Faull KF, Cuizon DB, Gage FH. Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J Cell Biol. 2001;153(2):283–294. doi:10.1083/jcb.153.2.283
23. Bae B, Xu H, Igarashi S, et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron. 2005;47(1):29–41. doi:10.1016/j.neuron.2005.06.005
24. Pryor WM, Biagioli M, Shahani N, et al. Huntingtin promotes mTORC1 signaling in the pathogenesis of Huntington’s disease. Sci Signal. 2014;7(349):1–13. doi:10.1126/scisignal.2005633
25. Gao R, Chakraborty A, Geater C, et al. Mutant Huntingtin impairs PNKP and ATXN3, disrupting DNA repair and transcription. Elife. 2019;8:1–31. doi:10.7554/eLife.42988
26. Martindale D, Hackam A, Wieczorek A, et al. Length of Huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat Genet. 1998;18(2):150–154. doi:10.1038/ng0298-150
27. Hackam AS, Singaraja R, Wellington CL, et al. The influence of Huntingtin protein size on nuclear localization and cellular toxicity. J Cell Biol. 1998;141(5):1097–1105. doi:10.1083/jcb.141.5.1097
28. Barbaro BA, Lukacsovich T, Agrawal N, et al. Comparative study of naturally occurring Huntingtin fragments in Drosophila points to exon 1 as the most pathogenic species in Huntington’s disease. Hum Mol Genet. 2015;24(4):913–925. doi:10.1093/hmg/ddu504
29. El‐Daher M, Hangen E, Bruyère J, et al. Huntingtin proteolysis releases non‐polyQ fragments that cause toxicity through dynamin 1 dysregulation. EMBO J. 2015;34(17):2255–2271. doi:10.15252/embj.201490808
30. Mangiarini L, Sathasivam K, Seller M, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87(3):493–506. doi:10.1016/S0092-8674(00)81369-0
31. Schilling G, Becher MW, Sharp AH, et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of Huntingtin. Hum Mol Genet. 1999;8(3):397–407. doi:10.1093/hmg/8.3.397
32. Tanaka Y, Igarashi S, Nakamura M, et al. Progressive phenotype and nuclear accumulation of an amino-terminal cleavage fragment in a transgenic mouse model with inducible expression of full-length mutant Huntingtin. Neurobiol Dis. 2006;21(2):381–391. doi:10.1016/j.nbd.2005.07.014
33. Landles C, Sathasivam K, Weiss A, et al. Proteolysis of mutant Huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease. J Biol Chem. 2010;285(12):8808–8823. doi:10.1074/jbc.M109.075028
34. Vieweg S, Mahul-Mellier AL, Ruggeri FS, et al. The Nt17 domain and its helical conformation regulate the aggregation, cellular properties and neurotoxicity of mutant Huntingtin exon 1. J Mol Biol. 2021;433(21):167222. doi:10.1016/j.jmb.2021.167222
35. Chongtham A, Bornemann DJ, Barbaro BA, et al. Effects of flanking sequences and cellular context on subcellular behavior and pathology of mutant HTT. Hum Mol Genet. 2020;29(4):674–688. doi:10.1093/hmg/ddaa001
36. Goldberg YP, Nicholson DW, Rasper DM, et al. Cleavage of Huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nat Genet. 1996;13(4):442–449. doi:10.1038/ng0896-442
37. Wellington CL, Ellerby LM, Hackam AS, et al. Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J Biol Chem. 1998;273(15):9158–9167. doi:10.1074/jbc.273.15.9158
38. Wellington CL, Singaraja R, Ellerby L, et al. Inhibiting caspase cleavage of Huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J Biol Chem. 2000;275(26):19831–19838. doi:10.1074/jbc.M001475200
39. Martin DDO, Schmidt ME, Nguyen YT, Lazic N, Hayden MR. Identification of a novel caspase cleavage site in huntingtin that regulates mutant huntingtin clearance. FASEB J. 2019;33(3):3190–3197. doi:10.1096/fj.201701510RRR
40. Kim YJ, Yi Y, Sapp E, et al. Caspase 3-cleaved N-terminal fragments of wild-type and mutant Huntingtin are present in normal and Huntington’s disease brains, associate with membranes, and undergo calpain dependent proteolysis. Proc Natl Acad Sci U S A. 2001;98(22):12784–12789. doi:10.1073/pnas.221451398
41. Gafni J, Ellerby LM. Calpain activation in Huntington’s disease. J Neurosci. 2002;22(12):4842–4849. doi:10.1523/JNEUROSCI.22-12-04842.2002
42. Sun B, Fan W, Balciunas A, et al. Polyglutamine repeat length-dependent proteolysis of Huntingtin. Neurobiol Dis. 2002;11(1):111–122. doi:10.1006/nbdi.2002.0539
43. Lunkes A, Lindenberg KS, Ben-Haem L, et al. Proteases acting on mutant Huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell. 2002;10(2):259–269. doi:10.1016/S1097-2765(02)00602-0
44. Schilling G, Klevytska A, Tebbenkamp ATN, et al. Characterization of Huntingtin pathologic fragments in human Huntington disease, transgenic mice, and cell models. J Neuropathol Exp Neurol. 2007;66(4):313–320. doi:10.1097/nen.0b013e318040b2c8
45. Tebbenkamp ATN, Crosby KW, Siemienski ZB, et al. Analysis of proteolytic processes and enzymatic activities in the generation of Huntingtin N-terminal fragments in an HEK293 cell model. PLoS One. 2012;7(12):1–10. doi:10.1371/journal.pone.0050750
46. Ratovitski T, Nakamura M, D’Ambola J, et al. N-terminal proteolysis of full-length mutant Huntingtin in an inducible PC12 cell model of Huntington’s disease. Cell Cycle. 2007;6(23):2970–2981. doi:10.4161/cc.6.23.4992
47. Sathasivam K, Neueder A, Gipson TA, et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci U S A. 2013;110(6):2366–2370. doi:10.1073/pnas.1221891110
48. Neueder A, Landles C, Ghosh R, et al. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci Rep. 2017;7(1):1–10. doi:10.1038/s41598-017-01510-z
49. Neueder A, Dumas AA, Benjamin AC, Bates GP. Regulatory mechanisms of incomplete Huntingtin mRNA splicing. Nat Commun. 2018;9(1). doi:10.1038/s41467-018-06281-3
50. Mason MA, Gomez-Paredes C, Sathasivam K, Neueder A, Papadopoulou AS, Bates GP. Silencing Srsf6 does not modulate incomplete splicing of the Huntingtin gene in Huntington’s disease models. Sci Rep. 2020;10(1):1–12. doi:10.1038/s41598-020-71111-w
51. Cooper JK, Schilling G, Peters MF, et al. Truncated N-terminal fragments of Huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum Mol Genet. 1998;7(5):783–790. doi:10.1093/hmg/7.5.783
52. Bates GP, Mangiarini L, Mahal A, Davies SW. Transgenic models of Huntington’s disease. Hum Mol Genet. 1997;6(10REV. ISS.):1633–1637. doi:10.1093/hmg/6.10.1633
53. Ramaswamy S, McBride JL, Kordower JH. Animal models of Huntington’s disease. ILAR J. 2007;48(4):356–373. doi:10.1093/ilar.48.4.356
54. Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell. 2000;101(1):57–66. doi:10.1016/S0092-8674(00
55. Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95(1):55–66. doi:10.1016/S0092-8674(00)81782-1
56. Hackam AS, Singaraja R, Zhang T, Gan L, Hayden MR. In vitro evidence for both the nucleus and cytoplasm as subcellular sites of pathogenesis in Huntington’s disease. Hum Mol Genet. 1999;8(1):25–33. doi:10.1093/hmg/8.1.25
57. Trushina E, Heldebrant MP, Perez-Terzic CM, et al. Microtubule destabilization and nuclear entry are sequential steps leading to toxicity in Huntington’s disease. Proc Natl Acad Sci U S A. 2003;100(21):12171–12176. doi:10.1073/pnas.2034961100
58. Wheeler VC, White JK, Gutekunst CA, et al. Long glutamine tracts cause nuclear localization of a novel form of Huntingtin in medium spiny striatal neurons in Hdh(Q92) and Hdh(Q111) knock-in mice. Hum Mol Genet. 2000;9(4):503–513. doi:10.1093/hmg/9.4.503
59. Lin CH, Tallaksen-Greene S, Chien WM, et al. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum Mol Genet. 2001;10(2):137–144. doi:10.1093/hmg/10.2.137
60. von Horsten S, Schmitt I, Nguyen HP, et al. Transgenic rat model of Huntington’s disease. Hum Mol Genet. 2003;12(6):617–624. doi:10.1093/hmg/ddg075
61. Slow EJ. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12(13):1555–1567. doi:10.1093/hmg/ddg169
62. Gray M, Shirasaki DI, Cepeda C, et al. Full-length human mutant Huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. 2008;28(24):6182–6195. doi:10.1523/JNEUROSCI.0857-08.2008
63. Jacobsen JC, Bawden CS, Rudiger SR, et al. An ovine transgenic Huntington’s disease model. Hum Mol Genet. 2010;19(10):1873–1882. doi:10.1093/hmg/ddq063
64. DiFiglia M, Sapp E, Chase KO, et al. Aggregation of Huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science (80-). 1997;277(5334):1990–1993. doi:10.1126/science.277.5334.1990
65. Anne Gutekunst C, Li SH, Yi H, et al. Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J Neurosci. 1999;19(7):2522–2534. doi:10.1523/JNEUROSCI.19-07-02522.1999
66. Sánchez I, Mahlke C, Yuan J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature. 2003;421(6921):373–379. doi:10.1038/nature01301
67. Schaffar G, Breuer P, Boteva R, et al. Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol Cell. 2004;15(1):95–105. doi:10.1016/j.molcel.2004.06.029
68. Takahashi T, Kikuchi S, Katada S, Nagai Y, Nishizawa M, Onodera O. Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum Mol Genet. 2008;17(3):345–356. doi:10.1093/hmg/ddm311
69. Leitman J, Hartl FU, Lederkremer GZ. Rather than large aggregates cause endoplasmic reticulum stress. Nat Commun. 2013;2013:1–10.
70. Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant Huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi:10.1038/nature02998
71. Chen JY, Parekh M, Seliman H, et al. Heat shock promotes inclusion body formation of mutant Huntingtin (mHtt) and alleviates mHtt-induced transcription factor dysfunction. J Biol Chem. 2018;293(40):15581–15593. doi:10.1074/jbc.RA118.002933
72. Arrasate M, Finkbeiner S. Protein aggregates in Huntington’s disease. Exp Neurol. 2012;238(1):1–11. doi:10.1016/j.expneurol.2011.12.013
73. Kim YE, Hosp F, Frottin F, et al. Soluble oligomers of PolyQ-expanded Huntingtin target a multiplicity of key cellular factors. Mol Cell. 2016;63(6):951–964. doi:10.1016/j.molcel.2016.07.022
74. Nucifora LG, Burke KA, Feng X, et al. Identification of novel potentially toxic oligomers formed in vitro from mammalian-derived expanded Huntingtin exon-1 protein. J Biol Chem. 2012;287(19):16017–16028. doi:10.1074/jbc.M111.252577
75. Kolla R, Gopinath P, Ricci J, Reif A, Rostami I, Lashuel HA. A new chemoenzymatic semisynthetic approach provides insight into the role of phosphorylation beyond exon1 of huntingtin and reveals N-terminal fragment length-dependent distinct mechanisms of aggregation. J Am Chem Soc. 2021;143(26):9798–9812. doi:10.1021/jacs.1c03108
76. Ehrnhoefer DE, Sutton L, Hayden MR. Small changes, big impact. Neurosci. 2011;17(5):475–492. doi:10.1177/1073858410390378
77. Lontay B, Kiss A, Virág L, Tar K. How do post-translational modifications influence the pathomechanistic landscape of Huntington’s disease? A comprehensive review. Int J Mol Sci. 2020;21(12):4282. doi:10.3390/ijms21124282
78. Aharony I, Ehrnhoefer DE, Shruster A, et al. A Huntingtin-based peptide inhibitor of caspase-6 provides protection from mutant Huntingtin-induced motor and behavioral deficits. Hum Mol Genet. 2015;24(9):2604–2614. doi:10.1093/hmg/ddv023
79. Leyva MJ, DeGiacomo F, Kaltenbach LS, et al. Identification and evaluation of small molecule pan-caspase inhibitors in Huntington’s disease models. Chem Biol. 2010;17(11):1189–1200. doi:10.1016/j.chembiol.2010.08.014
80. Graham RK, Deng Y, Slow EJ, et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant Huntingtin. Cell. 2006;125(6):1179–1191. doi:10.1016/j.cell.2006.04.026
81. Ona VO, Li M, Vonsattel JPG, et al. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington’s disease. Nature. 1999;399(6733):263–267. doi:10.1038/20446
82. Wang X, Zhu S, Drozda M, et al. Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington’s disease. Proc Natl Acad Sci. 2003;100(18):10483–10487. doi:10.1073/pnas.1832501100
83. Toulmond S, Tang K, Bureau Y, et al. Neuroprotective effects of M826, a reversible caspase-3 inhibitor, in the rat malonate model of Huntington’s disease. Br J Pharmacol. 2004;141(4):689–697. doi:10.1038/sj.bjp.0705662
84. Evers MM, Dai Tran H, Zalachoras I, et al. Preventing formation of toxic N-terminal Huntingtin fragments through antisense oligonucleotide-mediated protein modification. Nucleic Acid Ther. 2014;24(1):4–12. doi:10.1089/nat.2013.0452
85. Klein P, Karneva Z, Toonen L, et al. I01 QRX-704, a novel antisense oligonucleotide therapy, designed to prevent hd pathology while maintaining htt function. In: Experimental Therapeutics – Preclinical. BMJ Publishing Group Ltd; 2018:A88.2–A88. doi:10.1136/jnnp-2018-EHDN.237
86. Chaudhary RK, Patel KA, Patel MK, Joshi RH, Roy I. Inhibition of aggregation of mutant Huntingtin by nucleic acid aptamers in vitro and in a yeast model of Huntington’s disease. Mol Ther. 2015;23(12):1912–1926. doi:10.1038/mt.2015.157
87. Patel KA, Chaudhary RK, Roy I. RNA aptamers rescue mitochondrial dysfunction in a yeast model of Huntington’s disease. Mol Ther Nucleic Acid. 2018;12:45–56. doi:10.1016/j.omtn.2018.04.010
88. Messer A, Butler DC. Optimizing intracellular antibodies (intrabodies/nanobodies) to treat neurodegenerative disorders. Neurobiol Dis. 2020;134:104619. doi:10.1016/j.nbd.2019.104619
89. Butler DC, Snyder-Keller A, De Genst E, Messer A. Differential nuclear localization of complexes may underlie in vivo intrabody efficacy in Huntington’s disease. Protein Eng Des Sel. 2014;27(10):359–363. doi:10.1093/protein/gzu041
90. Southwell AL, Khoshnan A, Dunn DE, Bugg CW, Lo DC, Patterson PH. Intrabodies binding the proline-rich domains of mutant Huntingtin increase its turnover and reduce neurotoxicity. J Neurosci. 2008;28(36):9013–9020. doi:10.1523/JNEUROSCI.2747-08.2008
91. Southwell AL, Ko J, Patterson PH. Intrabody gene therapy ameliorates motor, cognitive, and neuropathological symptoms in multiple mouse models of Huntington’s disease. J Neurosci. 2009;29(43):13589–13602. doi:10.1523/JNEUROSCI.4286-09.2009
92. Southwell AL, Bugg CW, Kaltenbach LS, et al. Perturbation with intrabodies reveals that calpain cleavage is required for degradation of Huntingtin exon 1. PLoS One. 2011;6(1):e16676. doi:10.1371/journal.pone.0016676
93. De Genst E, Chirgadze DY, Klein FAC, et al. Structure of a single-chain Fv bound to the 17 N-terminal residues of Huntingtin provides insights into pathogenic amyloid formation and suppression. J Mol Biol. 2015;427(12):2166–2178. doi:10.1016/j.jmb.2015.03.021
94. Michel Lecerf J, Shirley TL, Zhu Q, et al. Human single-chain Fv intrabodies counteract in situ Huntingtin aggregation in cellular models of Huntington’s disease. Proc Natl Acad Sci. 2001;98(8):4764–4769. doi:10.1073/pnas.071058398
95. Murphy RC, Messer A. A single-chain Fv intrabody provides functional protection against the effects of mutant protein in an organotypic slice culture model of Huntington’s disease. Mol Brain Res. 2004;121(1–2):141–145. doi:10.1016/j.molbrainres.2003.11.011
96. Wolfgang WJ, Miller TW, Webster JM, et al. Suppression of Huntington’s disease pathology in Drosophila by human single-chain Fv antibodies. Proc Natl Acad Sci. 2005;102(32):11563–11568. doi:10.1073/pnas.0505321102
97. Miller TW, Zhou C, Gines S, et al. A human single-chain Fv intrabody preferentially targets amino-terminal Huntingtin fragments in striatal models of Huntington’s disease. Neurobiol Dis. 2005;19(1–2):47–56. doi:10.1016/j.nbd.2004.11.003
98. Snyder-Keller A, McLear JA, Hathorn T, Messer A. Early or late-stage anti-N-terminal Huntingtin intrabody gene therapy reduces pathological features in B6.HDR6/1 mice. J Neuropathol Exp Neurol. 2010;69(10):1078–1085. doi:10.1097/NEN.0b013e3181f530ec
99. Wang CE, Zhou H, McGuire JR, et al. Suppression of neuropil aggregates and neurological symptoms by an intracellular antibody implicates the cytoplasmic toxicity of mutant Huntingtin. J Cell Biol. 2008;181(5):803–816. doi:10.1083/jcb.200710158
100. Amaro IA, Henderson LA. An intrabody drug (rAAV6-INT41) reduces the binding of N-terminal Huntingtin fragment(s) to DNA to basal levels in PC12 cells and delays cognitive loss in the R6/2 animal model. J Neurodegener Dis. 2016;2016:1–10. doi:10.1155/2016/7120753
101. Jarosińska OD, Rüdiger SGD. Molecular strategies to target protein aggregation in Huntington’s disease. Front Mol Biosci. 2021;8:1–21. doi:10.3389/fmolb.2021.769184
102. Bauer PO, Goswami A, Wong HK, et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant Huntingtin protein. Nat Biotechnol. 2010;28(3):256–263. doi:10.1038/nbt.1608
103. Clift D, McEwan WA, Labzin LI, et al. A method for the acute and rapid degradation of endogenous proteins. Cell. 2017;171(7):1692–1706.e18. doi:10.1016/j.cell.2017.10.033
104. Butler DC, Messer A, Kahle PJ. Bifunctional anti-Huntingtin proteasome-directed intrabodies mediate efficient degradation of mutant Huntingtin exon 1 protein fragments. PLoS One. 2011;6(12):e29199. doi:10.1371/journal.pone.0029199
105. Ghosh B, Karmakar S, Prasad M, Mandal AK. Praja1 ubiquitin ligase facilitates degradation of polyglutamine proteins and suppresses polyglutamine-mediated toxicity. Mol Biol Cell. 2021;32(17):1579–1593. doi:10.1091/mbc.E20-11-0747
106. Hegde RN, Chiki A, Petricca L, et al. TBK1 phosphorylates mutant Huntingtin and suppresses its aggregation and toxicity in Huntington’s disease models. EMBO J. 2020;39(17):1–25. doi:10.15252/embj.2020104671
107. Aladdin A, Yao Y, Yang C, et al. The proteasome activators Blm10/PA200 enhance the proteasomal degradation of N-terminal Huntingtin. Biomolecules. 2020;10(11):1581. doi:10.3390/biom10111581
108. Galyan S, Ewald C, Jalencas X. Fragment-based virtual screening identifies a first- in-class preclinical drug candidate for Huntington ’ s disease. 2022:1–23.
109. Hu J, Matsui M, Gagnon KT, et al. Allele-specific silencing of mutant Huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs. Nat Biotechnol. 2009;27(5):478–484. doi:10.1038/nbt.1539
110. Hu J, Liu J, Corey DR. Allele-selective inhibition of Huntingtin expression by switching to an miRNA-like RNAi mechanism. Chem Biol. 2010;17(11):1183–1188. doi:10.1016/j.chembiol.2010.10.013
111. Gagnon KT, Pendergraff HM, Deleavey GF, et al. Allele-selective inhibition of mutant Huntingtin expression with antisense oligonucleotides targeting the expanded CAG repeat. Biochemistry. 2010;49(47):10166–10178. doi:10.1021/bi101208k
112. de Mezer M, Wojciechowska M, Napierala M, Sobczak K, Krzyzosiak WJ. Mutant CAG repeats of Huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucleic Acids Res. 2011;39(9):3852–3863. doi:10.1093/nar/gkq1323
113. Fiszer A, Mykowska A, Krzyzosiak WJ. Inhibition of mutant Huntingtin expression by RNA duplex targeting expanded CAG repeats. Nucleic Acids Res. 2011;39(13):5578–5585. doi:10.1093/nar/gkr156
114. Fiszer A, Ellison-Klimontowicz ME, Krzyzosiak WJ. Silencing of genes responsible for polyQ diseases using chemically modified single-stranded siRNAs. Acta Biochim Pol. 2016;63(4):759–764. doi:10.18388/abp.2016_1336
115. Yu D, Pendergraff H, Liu J, et al. Single-stranded RNAs use RNAi to potently and allele-selectively inhibit mutant Huntingtin expression. Cell. 2012;150(5):895–908. doi:10.1016/j.cell.2012.08.002
116. Aiba Y, Hu J, Liu J, Xiang Q, Martinez C, Corey DR. Allele-selective inhibition of expression of Huntingtin and ataxin-3 by RNA duplexes containing unlocked nucleic acid substitutions. Biochemistry. 2013;52(51):9329–9338. doi:10.1021/bi4014209
117. Liu J, Pendergraff H, Narayanannair KJ, et al. RNA duplexes with abasic substitutions are potent and allele-selective inhibitors of Huntingtin and ataxin-3 expression. Nucleic Acids Res. 2013;41(18):8788–8801. doi:10.1093/nar/gkt594
118. Monteys AM, Wilson MJ, Boudreau RL, Spengler RM, Davidson BL. Artificial miRNAs targeting mutant Huntingtin show preferential silencing in vitro and in vivo. Mol Ther. 2015;4:e234. doi:10.1038/mtna.2015.7
119. Urbanek MO, Fiszer A, Krzyzosiak WJ. Reduction of Huntington’s disease RNA Foci by CAG repeat-targeting reagents. Front Cell Neurosci. 2017;11:1–13. doi:10.3389/fncel.2017.00082
120. Datson NA, González-Barriga A, Kourkouta E, et al. The expanded CAG repeat in the Huntingtin gene as target for therapeutic RNA modulation throughout the HD mouse brain. Li Y, editor. PLoS One. 2017;12(2):e0171127. doi:10.1371/journal.pone.0171127
121. Ciesiolka A, Stroynowska-Czerwinska A, Joachimiak P, et al. Artificial miRNAs targeting CAG repeat expansion in ORFs cause rapid deadenylation and translation inhibition of mutant transcripts. Cell Mol Life Sci. 2021;78(4):1577–1596. doi:10.1007/s00018-020-03596-7
122. Kotowska-Zimmer A, Przybyl L, Pewinska M, et al. A CAG repeat-targeting artificial miRNA lowers the mutant Huntingtin level in the YAC128 model of Huntington’s disease. Mol Ther. 2022;28:702–715. doi:10.1016/j.omtn.2022.04.031
123. Miniarikova J, Zanella I, Huseinovic A, et al. Design, characterization, and lead selection of therapeutic miRNAs targeting Huntingtin for development of gene therapy for Huntington’s disease. Mol Ther. 2016;5:e297. doi:10.1038/mtna.2016.7
124. Miniarikova J, Zimmer V, Martier R, et al. AAV5-miHTT gene therapy demonstrates suppression of mutant Huntingtin aggregation and neuronal dysfunction in a rat model of Huntington’s disease. Gene Ther. 2017;24(10):630–639. doi:10.1038/gt.2017.71
125. Boado RJ, Kazantsev A, Apostol BL, Thompson LM, Pardridge WM. Antisense-mediated down-regulation of the human Huntingtin gene. J Pharmacol Exp Ther. 2000;295(1):239–243.
126. Chen ZJ, Kren BT, Wong PYP, Low WC, Steer CJ. Sleeping Beauty-mediated down-regulation of Huntingtin expression by RNA interference. Biochem Biophys Res Commun. 2005;329(2):646–652. doi:10.1016/j.bbrc.2005.02.024
127. Lai Wang Y, Liu W, Wada E, Murata M, Wada K, Kanazawa I. Clinico-pathological rescue of a model mouse of Huntington’s disease by siRNA. Neurosci Res. 2005;53(3):241–249. doi:10.1016/j.neures.2005.06.021
128. Rodriguez-Lebron E, Denovan-Wright EM, Nash K, Lewin AS, Mandel RJ. Intrastriatal rAAV-mediated delivery of anti-Huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington’s disease transgenic mice. Mol Ther. 2005;12(4):618–633. doi:10.1016/j.ymthe.2005.05.006
129. DiFiglia M, Sena-Esteves M, Chase K, et al. Therapeutic silencing of mutant Huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc Natl Acad Sci. 2007;104(43):17204–17209. doi:10.1073/pnas.0708285104
130. Denovan-Wright EM, Rodriguez-Lebron E, Lewin AS, Mandel RJ. Unexpected off-targeting effects of anti-Huntingtin ribozymes and siRNA in vivo. Neurobiol Dis. 2008;29(3):446–455. doi:10.1016/j.nbd.2007.11.003
131. Kordasiewicz HB, Stanek LM, Wancewicz EV, et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of Huntingtin synthesis. Neuron. 2012;74(6):1031–1044. doi:10.1016/j.neuron.2012.05.009
132. Evers MM, Miniarikova J, Juhas S, et al. AAV5-miHTT gene therapy demonstrates broad distribution and strong human mutant Huntingtin lowering in a Huntington’s disease minipig model. Mol Ther. 2018;26(9):2163–2177. doi:10.1016/j.ymthe.2018.06.021
133. Caron NS, Southwell AL, Brouwers CC, et al. Potent and sustained Huntingtin lowering via AAV5 encoding miRNA preserves striatal volume and cognitive function in a humanized mouse model of Huntington disease. Nucleic Acids Res. 2019;48(1):36–54. doi:10.1093/nar/gkz976
134. Spronck EA, Brouwers CC, Vallès A, et al. AAV5-miHTT gene therapy demonstrates sustained Huntingtin lowering and functional improvement in Huntington disease mouse models. Mol Ther. 2019;13:334–343. doi:10.1016/j.omtm.2019.03.002
135. Keskin S, Brouwers CC, Sogorb-Gonzalez M, et al. AAV5-miHTT lowers Huntingtin mRNA and protein without off-target effects in patient-derived neuronal cultures and astrocytes. Mol Ther. 2019;15:275–284. doi:10.1016/j.omtm.2019.09.010
136. Vallès A, Evers MM, Stam A, et al. Widespread and sustained target engagement in Huntington’s disease minipigs upon intrastriatal microRNA-based gene therapy. Sci Transl Med. 2021;13(588):1–13. doi:10.1126/scitranslmed.abb8920
137. Spronck E, Vallès A, Lampen M, et al. Intrastriatal administration of AAV5-miHTT in non-human primates and rats is well tolerated and results in miHTT transgene expression in key areas of Huntington disease pathology. Brain Sci. 2021;11(2):129. doi:10.3390/brainsci11020129
138. Rindt H, Yen PF, Thebeau CN, Peterson TS, Weisman GA, Lorson CL. Replacement of Huntingtin exon 1 by trans-splicing. Cell Mol Life Sci. 2012;69(24):4191–4204. doi:10.1007/s00018-012-1083-5
139. Rindt H, Tom CM, Lorson CL, Mattis VB. Optimization of trans-splicing for Huntington’s disease RNA therapy. Front Neurosci. 2017;11:1–13. doi:10.3389/fnins.2017.00544
140. Batra R, Nelles DA, Pirie E, et al. Elimination of toxic microsatellite repeat expansion RNA by RNA-targeting Cas9. Cell. 2017;170(5):899–912.e10. doi:10.1016/j.cell.2017.07.010
141. Batra R, Nelles DA, Roth DM, et al. The sustained expression of Cas9 targeting toxic RNAs reverses disease phenotypes in mouse models of myotonic dystrophy type 1. Nat Biomed Eng. 2021;5(2):157–168. doi:10.1038/s41551-020-00607-7
142. Garriga-Canut M, Agustín-Pavón C, Herrmann F, et al. Synthetic zinc finger repressors reduce mutant Huntingtin expression in the brain of R6/2 mice. Proc Natl Acad Sci. 2012;109(45). doi:10.1073/pnas.1206506109
143. Agustín-Pavón C, Mielcarek M, Garriga-Canut M, Isalan M. Deimmunization for gene therapy: host matching of synthetic zinc finger constructs enables long-term mutant Huntingtin repression in mice. Mol Neurodegener. 2016;11(1):64. doi:10.1186/s13024-016-0128-x
144. Zeitler B, Froelich S, Marlen K, et al. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat Med. 2019;25(7):1131–1142. doi:10.1038/s41591-019-0478-3
145. Kolli N, Lu M, Maiti P, Rossignol J, Dunbar G. CRISPR-Cas9 mediated gene-silencing of the mutant Huntingtin gene in an in vitro model of Huntington’s disease. Int J Mol Sci. 2017;18(4):754. doi:10.3390/ijms18040754
146. Merienne N, Vachey G, de Longprez L, et al. The self-inactivating KamiCas9 system for the editing of CNS disease genes. Cell Rep. 2017;20(12):2980–2991. doi:10.1016/j.celrep.2017.08.075
147. Ekman FK, Ojala DS, Adil MM, Lopez PA, Schaffer DV, Gaj T. CRISPR-Cas9-mediated genome editing increases lifespan and improves motor deficits in a Huntington’s disease mouse model. Mol Ther. 2019;17:829–839. doi:10.1016/j.omtn.2019.07.009
148. Powell JE, Lim CKW, Krishnan R, et al. Targeted gene silencing in the nervous system with CRISPR-Cas13. Sci Adv. 2022;8(3):1–11. doi:10.1126/sciadv.abk2485
149. Shin JW, Hee Kim K, Chao MJ, et al. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum Mol Genet. 2016;25(20):ddw286. doi:10.1093/hmg/ddw286
150. Yang S, Chang R, Yang H, et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J Clin Invest. 2017;127(7):2719–2724. doi:10.1172/JCI92087
151. Monteys AM, Ebanks SA, Keiser MS, Davidson BL. CRISPR/Cas9 editing of the mutant Huntingtin allele in vitro and in vivo. Mol Ther. 2017;25(1):12–23. doi:10.1016/j.ymthe.2016.11.010
152. Dabrowska M, Juzwa W, Krzyzosiak WJ, Olejniczak M. Precise excision of the CAG tract from the Huntingtin gene by Cas9 nickases. Front Neurosci. 2018;12:1–8. doi:10.3389/fnins.2018.00075
153. Wu J, Tang Y, Li Zhang C. Targeting N-terminal Huntingtin with a dual-sgRNA strategy by CRISPR/Cas9. Biomed Res Int. 2019;2019:1–10. doi:10.1155/2019/1039623
154. Lopes C, Tang Y, Anjo SI, et al. Mitochondrial and redox modifications in Huntington disease induced pluripotent stem cells rescued by CRISPR/Cas9 CAGs targeting. Front Cell Dev Biol. 2020;8:1–19. doi:10.3389/fcell.2020.576592
155. Rook ME, Southwell AL. Antisense oligonucleotide therapy: from design to the Huntington disease clinic. BioDrugs. 2022;36(2):105–119. doi:10.1007/s40259-022-00519-9
156. Bhattacharyya A, Trotta CR, Narasimhan J, et al. Small molecule splicing modifiers with systemic HTT-lowering activity. Nat Commun. 2021;12(1):7299. doi:10.1038/s41467-021-27157-z
157. Keller CG, Shin Y, Monteys AM, et al. An orally available, brain penetrant, small molecule lowers Huntingtin levels by enhancing pseudoexon inclusion. Nat Commun. 2022;13(1):1–11. doi:10.1038/s41467-022-28653-6
158. Arnoux I, Willam M, Griesche N, et al. Metformin reverses early cortical network dysfunction and behavior changes in Huntington’s disease. Elife. 2018;7:1–32. doi:10.7554/eLife.38744
159. Goertsen D, Flytzanis NC, Goeden N, et al. AAV capsid variants with brain-wide transgene expression and decreased liver targeting after intravenous delivery in mouse and marmoset. Nat Neurosci. 2022;25(1):106–115. doi:10.1038/s41593-021-00969-4
160. Scahill RI, Zeun P, Osborne-Crowley K, et al. Biological and clinical characteristics of gene carriers far from predicted onset in the Huntington’s disease Young Adult Study (HD-YAS): a cross-sectional analysis. Lancet Neurol. 2020;19(6):502–512. doi:10.1016/S1474-4422(20)30143-5
161. Miller JP, Holcomb J, Al-Ramahi I, et al. Matrix metalloproteinases are modifiers of Huntingtin proteolysis and toxicity in Huntington’s Ddsease. Neuron. 2010;67(2):199–212. doi:10.1016/j.neuron.2010.06.021
162. Ratovitski T, Gucek M, Jiang H, et al. Mutant Huntingtin N-terminal fragments of specific size mediate aggregation and toxicity in neuronal cells. J Biol Chem. 2009;284(16):10855–10867. doi:10.1074/jbc.M804813200
163. Ratovitski T, Chighladze E, Waldron E, Hirschhorn RR, Ross CA. Cysteine proteases bleomycin hydrolase and cathepsin Z mediate N-terminal proteolysis and toxicity of mutant Huntingtin. J Biol Chem. 2011;286(14):12578–12589. doi:10.1074/jbc.M110.185348
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.