")
Back to Journals » Research and Reports in Urology » Volume 10
Enzalutamide and blocking androgen receptor in advanced prostate cancer: lessons learnt from the history of drug development of antiandrogens
Received 15 November 2017
Accepted for publication 13 January 2018
Published 16 February 2018 Volume 2018:10 Pages 23—32
DOI https://doi.org/10.2147/RRU.S157116
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jan Colli
Yusuke Ito, Marianne D Sadar
Genome Sciences Centre, BC Cancer, Vancouver, BC, Canada
Abstract: Enzalutamide is a nonsteroidal antiandrogen for the treatment of metastatic castration-resistant prostate cancer (mCRPC) both before and after chemotherapy. Enzalutamide is more effective than its predecessor bicalutamide, which was analyzed in head-to-head studies of patients with CRPC. This family of nonsteroidal antiandrogens is now comprised of four drugs approved by the US Food and Drug Administration with two investigational drugs in clinical trials. Antiandrogens have been employed clinically for more than five decades to provide a rich resource of information. Steady-state concentration minimums (Cmin or trough) in the range of ~1–13 μg/mL are measured in patients at therapeutic doses. Interestingly, enzalutamide which is considered to have strong affinity for the androgen receptor (AR) requires Cmin levels >10 μg/mL. The sequence of antiandrogens and the clinical order of application in regard to other drugs that target the androgen axis remain of high interest. One novel first-in-class drug, called ralaniten, which binds to a unique region in the N-terminus domain of both the full-length and the truncated constitutively active splice variants of the AR, is currently in clinical trials for patients who previously received abiraterone, enzalutamide, or both. This highlights the trend to develop drugs with novel mechanisms of action and potentially differing mechanisms of resistance compared with antiandrogens. Better and more complete inhibition of the transcriptional activity of the AR appears to continue to provide improvements in the clinical management of mCRPC.
Keywords: prostate cancer, enzalutamide, antiandrogens, Cmin, trough, pharmacology, cross-resistance, clinical trials, PSA response, ralaniten, EPI-002
Introduction
Prostate cancer is the most commonly diagnosed cancer in men in the Western world. An estimated 26,730 men were predicted to die of prostate cancer in 2017 in the USA.1 The terminal stage of prostate cancer is called metastatic castration-resistant prostate cancer (mCRPC). Most prostate cancers and stages of prostate cancer depend upon androgen and the androgen receptor (AR) for their growth and survival. This dependency upon androgen (hormone) makes prostate cancer distinct from nonhormonal malignancies.
The AR is a transcription factor that regulates the expression of hundreds of genes in response to binding androgen. In other words, the AR mediates the effects of androgens such as testosterone and its more active metabolite dihydrotestosterone by altering the levels of expression of genes involved in proliferation and survival. Systemic treatment for advanced prostate cancer involves androgen deprivation therapy (ADT) that is applied to prevent the transcriptional activity of the AR. There are two main therapeutic approaches used in ADT: the first is to reduce the levels of circulating androgen by either surgical or chemical castration, and the second is to prevent androgen from binding to the AR by the application of a competitive inhibitor called antiandrogen. Reduction of circulating levels of androgen by >90% within 24 hours is achieved by surgical castration.2 Chemical castration applies analogs of luteinizing hormone–releasing hormone (LH-RH) and is comparable to surgical castration in reducing circulating levels of testosterone. LH-RH agonists include leuprolide acetate and goserelin acetate, while degarelix is an antagonist. Nonsteroidal, first-generation antiandrogens include flutamide, nilutamide, and bicalutamide. LH-RH agonists and antiandrogens have been the front line of hormone therapy for advanced prostate cancer, but this therapy is not curative. New treatments for CRPC include more potent hormone therapies such as the second-generation antiandrogen enzalutamide and the CYP17 inhibitor, abiraterone acetate, which prevents the synthesis of androgens. However, these too will eventually fail and require other therapies to be administered. Nonhormonal therapies approved for mCRPC include taxanes, sipuleucel-T (immunotherapy), and radium-223. Unfortunately, these nonhormonal therapies only increase survival time by several months with patients succumbing to mCRPC. Most mCRPC is still considered to be driven by a transcriptionally active AR in spite of castrate levels of circulating androgens. Thus, the search for novel approaches to block the transcriptional activity of the AR remains the focus of current drug development programs, of which the antiandrogen enzalutamide is an example that we highlight against the background of other antiandrogens and new investigational drugs that directly target the AR.
Overview of current antiandrogen treatment options
Cyproterone acetate
The drug development of antiandrogens began in 1962 with the steroidal antiandrogens, which have a steroidal chemical structure. These are progestogens and include cyproterone acetate, chlormadinone acetate, megestrol acetate, and dienogest. Of these, cyproterone acetate (Figure 1) is the most potent steroidal antiandrogen and is used for the treatment of prostate cancer. It is a derivative of hydroxyprogesterone that has a better relative binding affinity for AR than any of the first-generation, nonsteroidal antiandrogens.3,4 However, in vitro binding assays may not be predictive of in vivo antiandrogenic efficacy5 and differ depending upon whether the experiment is performed in castrated or intact animals.6 Cyproterone acetate is both an antigonadotropin and an antiandrogen that competes with androgen for the ligand-binding domain of AR. It is not a pure antagonist, but rather is a partial agonist. These are important differences between steroidal antiandrogens and nonsteroidal antiandrogens that impact their therapeutic use. Unlike nonsteroidal antiandrogens, cyproterone acetate does not increase the survival of prostate cancer patients when combined with castration.7 Rather, cyproterone acetate adversely affected survival.8 Cyproterone acetate is effective in preventing flare responses that occur with chemical castration of prostate cancer patients treated with LH-RH agonists. Its terminal half-life is ~38 hours in plasma.
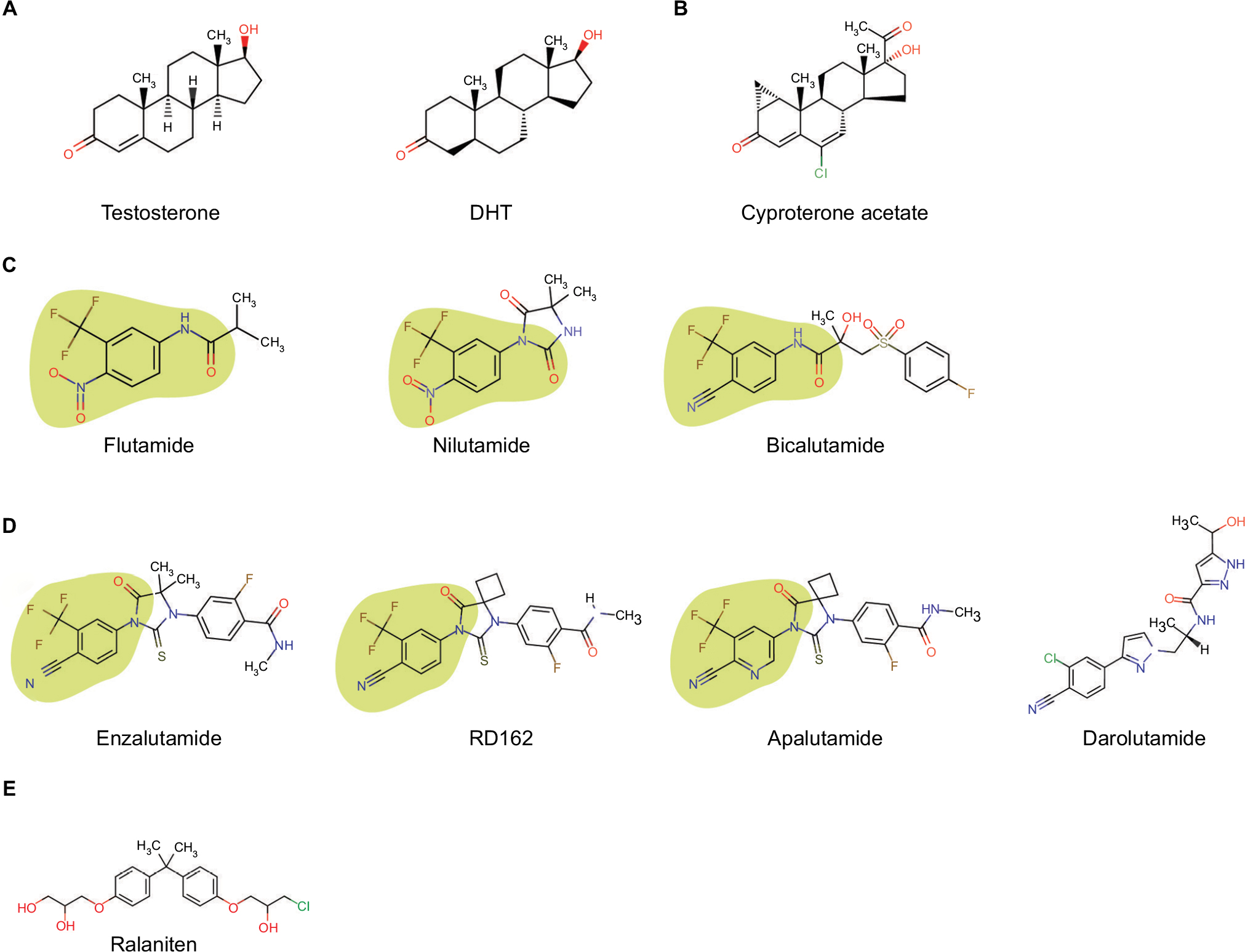 | Figure 1 Compounds that bind the androgen receptor. Notes: (A) Testosterone and DHT are androgens and agonists of the androgen receptor; (B) steroidal antiandrogen, cyproterone acetate, which shares some of the steroid chemical structure of androgens; (C) first-generation nonsteroidal antiandrogens that do not have a steroid chemical structure; (D) second-generation nonsteroidal antiandrogens that share some aspects of the chemical structure of first-generation nonsteroidal antiandrogens with the exception of darolutamide; (E) N-terminal domain antagonist ralaniten. Note: The green area indicates the common chemical moiety of nonsteroidal antiandrogens that bind in the ligand-binding pocket of the androgen receptor. Abbreviation: DHT, dihydrotestosterone. |
Flutamide
Flutamide was the first nonsteroidal antiandrogen approved by the US Food and Drug Administration (FDA) for prostate cancer. This first-in-class drug still forms the structural basis of other nonsteroidal antiandrogens including enzalutamide and apalutamide (Figure 1). The discovery of a chemical moiety that binds in the ligand-binding pocket of the AR was a major achievement in drug development for prostate cancer, and it continues to be used to target the full-length AR such as those with proteolysis-targeting chimeras AR degraders and selective AR modulators.9,10 Structure–activity relationship studies have been described for nonsteroidal ligands with variations in this chemical moiety.11–13
Flutamide is a pure antiandrogen and was considered to not have AR agonist activity nor any of the progestational adverse effects of the steroidal antiandrogens. The mechanism of action is reported to block binding of androgen to the ligand-binding pocket of the AR as well as to decrease nuclear translocation of androgen-bound AR.14 Gain-of-function mutations in the ligand-binding domain of AR, such as T877A, have been identified in patients whose disease improves upon cessation of treatment with flutamide.15,16 This phenomenon has been termed “antiandrogen withdrawal syndrome” and is suggested to happen with all antiandrogens including enzalutamide and the investigational agent apalutamide. Upon first-pass metabolism, flutamide is metabolized to its more active metabolite, 2-hydroxyflutamide, as well as a potentially toxic hydrolysis product, 3-trifluormethyl-4-nitroaniline. The elimination route of active metabolites is via the kidneys. Liver toxicity is one of the most common adverse effects from flutamide. This together with its short half-life of ~6 hours17,18 limited its usage compared with other available nonsteroidal antiandrogens. The recommended dose of flutamide is 250 mg three times per day to give Cmax and Cmin of ~1.7 and 0.8 μg/mL, respectively.19 Indications for flutamide are in combination with LH-RH agonists in locally confined stage B2-C and stage D2 metastatic prostate carcinoma.
Nilutamide
Nilutamide is approved as a pure nonsteroidal antiandrogen for stage D2 metastatic prostate cancer in combination with castration. The therapeutic activity of nilutamide is attributed to the parent compound. Its half-life is ~56 hours.20 After an initial dose of 300 mg per day for 30 days starting the day of or the day after surgical castration, the dose is reduced to 150 mg once daily. Cmin at steady state is 6–7 μg/mL for a dose of 300 mg/day.21 Metabolism of nilutamide is by reduction of the nitro group and amino derivative, which are excreted predominately in the urine. Contraindications include severe hepatic impairment or respiratory insufficiency. Antiandrogen withdrawal responses have also been observed in some patients who stop nilutamide treatment which include a drop in levels of serum prostate-specific antigen (PSA).22,23
Bicalutamide
Bicalutamide has had the most widespread application as a nonsteroidal antiandrogen for prostate cancer. It is administered as a mixture of stereoisomers in spite of the fact that the R-isomer has 30-fold better binding affinity compared with the S-isomer.24 Bicalutamide is FDA-approved at 50 mg daily in combination with LH-RH analog for prostate cancer patients with stage D2 disease. Cmin is ~9.33 μg/mL after 12 weeks of dosing.25 However, higher doses led to increased Cmin values that plateau at ~30 μg/mL with doses between 150 and 200 mg/day.26 A trend in reduction of total PSA with increased dosage also plateaued at 150/200 mg/day,27 thereby suggesting that 50 mg per day of bicalutamide is suboptimal. Compared with flutamide and nilutamide, bicalutamide has less hepatotoxicity and a longer half-life of ~6 days.26 Bicalutamide withdrawal responses have also been observed28–30 and are suggested to be involved in mutations in the ligand-binding domain of the AR such as W741C/L.31,32 Similar to enzalutamide, bicalutamide is also a potent inhibitor of the structurally related progesterone receptor.33–35
Clinical application of flutamide, nilutamide, and bicalutamide for CRPC has yielded some benefits. High-dose bicalutamide (150 mg/day) resulted in a decline of serum PSA in 44.7% of patients with a duration of response of >1.5 years,36 which is consistent with better Cmin values reported for this dose.26 Similarly, 29% of patients who received nilutamide had a sustained reduction in serum PSA beyond 3 months,37 whereas 50% of 16 patients with CRPC who were treated with 375 mg/day flutamide achieved a decline in serum PSA of >50%.38 Sequential application of nonsteroidal antiandrogens leads to a PSA response rate of ~36% (≥50% drop in serum PSA) in a 6.6-month duration.39 Whereas switching to a third-line antiandrogen is less effective with a response rate of merely 13%–29%.39–41 PSA response for first-line antiandrogen therapy or PSA levels <3 ng/mL at the start of second-line antiandrogen therapy were significantly related to the response to second-line antiandrogen therapy.39,42,43 Collectively, these studies showed that responses to second-line antiandrogens were achievable with significant increases in survival.39–42 These studies also led to the development of second-generation antiandrogens such as FDA-approved enzalutamide and perhaps foreshadowed the cross-resistance that would be observed more than one decade later with more potent inhibitors such as abiraterone acetate and enzalutamide, when used sequentially.
Enzalutamide
Enzalutamide (previously called MDV3100) was screened from ~200 synthesized nonsteroidal antiandrogens using lymph node carcinoma of the prostate (LNCaP) cells as a model.44 Two lead compounds, RD162 and enzalutamide (Figure 1), were pursued, and enzalutamide was selected based upon activity and favorable drug-like properties. Enzalutamide inhibits the following: binding of androgens to the ligand-binding domain of AR; nuclear translocation of AR; binding of AR to DNA; and interactions of AR with coactivators.44 Although enzalutamide is chemically related to bicalutamide (Figure 1), it has five to eight times better affinity for AR based upon competitive displacement of 18F-16b-fluoro-5a-dihydrotestosterone (18F-FDHT) equilibrium binding to the AR.44 Unlike bicalutamide, enzalutamide does not have an agonistic effect on the transcriptional activity of the AR. However, gain-of-function mutations in the AR ligand-binding domain (eg, F876L) and the expression of constitutively active splice variants of the AR that lack a ligand-binding domain are considered to be the main mechanisms of clinical resistance.
Enzalutamide is primarily eliminated by hepatic metabolism by CYP3A4 and CYP2C8, which raises concerns about drug–drug interactions compared with competing compounds such as abiraterone acetate. Its major metabolite, N-desmethyl enzalutamide, is formed by CYP2B8 and has similar in vitro activity compared with enzalutamide. Enzalutamide and N-desmethyl enzalutamide have half-lives of 5.8 and 8.6 days with corresponding Cmin values of 11.4 and 13 μg/mL, respectively, when taking a daily oral dose of 160 mg in combination with LH-RH analogs. Cmin levels between 5 and 15 μg/mL of enzalutamide are suggested to be required to saturate binding to the AR.45 Dose escalation studies showed that the magnitude of reduction of serum PSA and the proportion of patients with a PSA response were dose-dependent.45 The recommended dose of 160 mg/day was within a plateau observed between 150 and 240 mg per day.45
Enzalutamide was FDA-approved in 2012 for the treatment of patients with mCRPC who previously received docetaxel based upon the results from the AFFIRM phase III clinical study. Later, enzalutamide was approved for chemo-naïve patients based upon the PREVAIL trial.46 The PSA response rates of >50% PSA reduction were 54% in the AFFIRM trial and 78% in the PREVAIL trial. Lesser PSA response rates were observed in those patients who previously received abiraterone acetate plus prednisone, which suggests that clinical cross-resistance occurs in spite of different mechanisms of action within the androgen axis.47 Of 214 patients who previously received abiraterone therapy, only 27% (48 of 181 patients) had a PSA response.47 The median time to PSA progression was 5.7 months, whereas the median radiographic progression-free survival (PFS) was 8.1 months.47 Retrospective analyses have reported similar reduced response rates in those patients who received enzalutamide after progression on abiraterone.48,49 These data are reminiscent of the previous clinical studies examining the sequence of first-generation antiandrogens. Importantly, it emphasizes that there is still no gold standard for the sequential order of enzalutamide and abiraterone acetate. Currently, these two drugs are prescribed according to patient past history and condition. We hypothesize that the mechanism of resistance observed with enzalutamide subsequent to abiraterone treatment may involve the conversion of abiraterone to Δ4-abiraterone which acts as an antiandrogen.50 Although abiraterone acetate plus prednisone therapy involves three mechanisms that are specifically CYP17A, AR, and glucocorticoid receptor, there is some support showing that abiraterone-to-enzalutamide sequence of application has better efficacy compared with enzalutamide-to-abiraterone sequence.51 Unlike abiraterone acetate, enzalutamide can be taken regardless of food, has mild hepatic impairment, and does not need prednisone.
Current developments in investigational new drugs
The success of enzalutamide in the treatment of mCRPC together with the general acceptance that a transcriptionally active AR still drives this stage of the disease emphasizes the need for additional approaches to block the activity of this transcription factor. Several investigational drugs are currently in clinical trials that also target the AR either as an antiandrogen or through a more novel mechanism involving the amino-terminal domain instead of the C-terminal ligand-binding domain of this receptor.
Apalutamide
Apalutamide is a second-generation, nonsteroidal antiandrogen that differs from RD162 by one atom and from enzalutamide by two atoms (Figure 1). As predicted based upon similar chemical structure, many properties are similar between enzalutamide and apalutamide, including that both are agonists for the mutated AR (F876L)52 and have comparable IC50s for competitive inhibition of 18F-FDHT binding to the AR (16.0±2.1 nM vs 21.4±4.4 nM).53 However, differences in AR-binding assays were dependent on the cell line used with no differences observed between bicalutamide and apalutamide in MDA-MB-453 cells in spite of the 10-fold difference found in LNCaP cells.53 Substantial differences in pharmacokinetic parameters in preclinical studies were measured with enzalutamide having almost twice as long plasma half-life compared with apalutamide (ie, 15.8 hours vs 8.7 hours, respectively, at doses of 10 mg/kg).53 Thus, corresponding Cmax levels after 42 days of dosing in mice at 10 mg/kg were 34.4 μg/mL versus 9.66 μg/mL and Cmin levels were 13.8 μg/mL versus 3.91 μg/mL between enzalutamide and apalutamide, respectively. Curiously, in spite of this ~3-fold difference in plasma levels of drug in mice, negligible differences in drug concentrations were measured in the harvested xenografts between the two related drugs, both at ~3.3 μg/g tissue at steady state.53 This was speculated to possibly be due to differences in the steady-state volume of distribution and significant differences in protein-binding, with apalutamide having at least 2-fold greater free fraction compared with enzalutamide in mouse and human plasma.53 Phase I clinical data determined a recommended phase II dose of 240 mg per day based upon Cmin trough levels in patients ranging between 3 and 6 μg/mL, which were required in preclinical studies in mice to elicit the regression of LNCaP xenografts.54 The phase II dose of 240 mg per day gave a half-life of 86.2 hours, a steady-state Cmax of 7.6 μg/mL, and Cmin of ~4 μg/mL.55 Using FDHT-positron emission tomography/computed tomography imaging to assess a pharmacodynamic response, a plateau was obtained at a Cmin of ≥2μg/mL using a ≥120 mg day dose, which was interpreted to be the concentration needed to fully occupy the available AR-binding sites.55 Apalutamide at 240 mg/day showed diminished activity in patients who had received abiraterone acetate compared with those with no prior abiraterone acetate treatment.56 The 12-week PSA response rate was 22% versus 88%, median time to PSA progression was 3.7 months versus 18.2 months, and median time on treatment was 4.9 months versus 21 months for patients who previously received abiraterone acetate versus those who had not had abiraterone therapy.56 Importantly, this trial highlighted the disconnect between time on therapy and the degree of PSA decline. Forty-three percent of patients who had previously received abiraterone therapy remained on apalutamide therapy for ≥6 months.56 The authors conclude that a ≥50% decline in PSA as an indicator of a “favorable treatment effect” can underestimate the proportion of patients who could be receiving benefit,56 consistent with others.57,58 This seems to be especially of importance for therapies that target a similar pathway such as the androgen axis. In phase II clinical study of high-risk nonmetastatic CRPC, 240 mg per day of apalutamide showed that 89% of patients had ≥50% PSA decline at 12 weeks.59 These studies are being expanded in the SPARTAN multicenter double-blind phase III clinical trial to evaluate the efficacy and safety of apalutamide compared with placebo-control in 1200 high-risk nonmetastatic CRPC (m0CRPC) patients (NCT01946204). A phase III study using a combination of apalutamide with abiraterone plus prednisone is also ongoing (NCT02257736).
Darolutamide
Darolutamide is an oral nonsteroidal antiandrogen that is a mixture of two diastereomers (ORM-16497 and ORM-16555). The main metabolite, ORM-15341, is potent with more than 10× lower inhibition constant compared with enzalutamide or apalutamide in a competitive AR-binding assay.60 These structures of darolutamide are distinct from the structures of other nonsteroidal antiandrogens (Figure 1), which may result in differences in biology and resistant mechanisms. One such mechanism is the emergence of gain-of-function mutations in the AR ligand-binding domain that occurs with all of the current antiandrogens. Darolutamide blocks the activities of these known mutant ARs including F876L, which confers resistance to both enzalutamide and apalutamide. Darolutamide has negligible blood–brain barrier penetration compared with other nonsteroidal antiandrogens and does not elevate levels of testosterone. Phase I ARAFOR trial61 and phase I/II ARADES trial62 showed a favorable safety profile. Steady-state concentrations for doses of 1000–1400 mg per day ranged between ~1.8 and 4.3 μg/mL, respectively.62 The mean terminal half-lives of darolutamide and its active metabolite ORM-15341 were independent of dose and at steady state were 15.8 and 10.0 hours, respectively.62 Based upon these half-lives, two ongoing global double-blind phase III trials, ARAMIS (NCT02200614) in nonmetastatic CRPC and ARASENS (NCT02799602) in metastatic hormone-sensitive prostate cancer, are using 1200 mg/day dosed at 600 mg twice a day.
Ralaniten acetate
EPI analogs such as ralaniten (formerly known as EPI-002) are the first class of small molecules to be discovered that bind to the intrinsically disordered N-terminus of the AR.63–67 They also represent the first small molecules proven to bind to the intrinsically disordered N-terminal domain of any steroid hormone receptor. This is of particular importance in the field of prostate cancer because the disease is driven by the AR and, 1) the transcriptional activity of the AR resides in the N-terminal domain; and 2) the loss or truncation of the C-terminal ligand-binding domain yields constitutively active ARs. Thus, a small molecule that binds to critical regions in the N-terminal domain of the AR would block the transcriptional activities of both the full-length and truncated splice variants of the AR. Indeed, the EPI compounds have been shown in preclinical models to block the transcriptional activities of full-length AR, AR-V7, AR-V567es, as well as gain-of-function mutations of the AR.65,68 The expression of constitutively active AR-V7 in circulating tumor cells has been reported as a biomarker of enzalutamide and abiraterone resistance in mCRPC.69 Recently, the prodrug of ralaniten called ralaniten acetate or EPI-506 started phase I dose escalation clinical trials in patients who were previously treated with enzalutamide, abiraterone acetate, or both (NCT02606123).63,65,70 The fact that this is the first clinical trial for any drug that targets an intrinsically disordered region is important in the field of drug development.71
Comparative efficacy, safety, and tolerability of enzalutamide
The TERRAIN trial72 and the STRIVE73 trial are the most useful clinical studies that directly compare the efficacy and adverse effects of enzalutamide versus bicalutamide. TERRAIN was a double-blind, randomized, phase II study that evaluated the efficacy and adverse effects of enzalutamide (160 mg/day) versus bicalutamide (50 mg/day) in mCRPC. The primary endpoint was PFS, which was significantly improved in the enzalutamide group (median PFS of 15.7 months) compared with patients in the bicalutamide group (5.8 months). STRIVE was also a double-blind, randomized phase II study and employed the same dose of enzalutamide (160 mg/day) and bicalutamide (50 mg/day); however, it included not only mCRPC but also m0CRPC patients. The median PFS was significantly improved in the enzalutamide group (19.4 months) compared with patients in the bicalutamide group (5.7 months). The major weakness of the trial was the caution that 50 mg per day of bicalutamide may have been a suboptimal dose and does not globally reflect clinical practice.
The most common adverse events of enzalutamide reported in the AFFIRM, PREVAIL, TERRAIN, and STRIVE clinical studies were fatigue, back pain, and hot flushes. Seizure is a risk with enzalutamide treatment; therefore, patients with a history of seizure were excluded from the more recent studies. Post hoc analyses of both the AFFIRM and PREVAIL trials showed that enzalutamide was generally well tolerated and effective even in the elderly subgroup aged >75 years.74 The increased occurrence of falls in elderly patients with enzalutamide treatment (19.2%) compared with placebo (7.9%) was of note.74
No head-to-head study has compared the efficacy between enzalutamide versus abiraterone acetate plus prednisone regardless of pre- or postchemotherapy setting. Some groups reported the indirect comparison analysis of these drugs using the clinical data from the PREVAIL, AFFIRM, and COU-AA-301/302 studies.75,76 According to these reports, there is no strong evidence that enzalutamide is superior to abiraterone plus prednisone for overall survival. However, there is strong evidence that enzalutamide surpasses abiraterone acetate plus prednisone in terms of secondary endpoints that include PFS, time to PSA progression, and PSA response rate regardless of pre- or postchemotherapy setting.
Patient-focused perspectives such as quality of life, satisfaction, acceptability, and adherence
Patient-reported outcome is an important endpoint that should be evaluated in clinical studies. This is because medical doctors may underestimate adverse events, or there may be an inconsistency between patients and doctors about the recognition of health-related quality of life.77 The subjectivity of patients’ reports makes evaluation challenging. Fortunately, some tools and questionnaires are now available. The Functional Assessment of Cancer Therapy – General (FACT-G) was the original questionnaire used to assess health-related quality of life in patients receiving cancer therapy. The Functional Assessment of Cancer Therapy – Prostate (FACT-P) is a questionnaire based upon FACT-G that includes 12 questions measuring prostate cancer-specific factors affecting the quality of life. A recent review has examined the impact of enzalutamide on patient-reported outcomes.78
In the TERRAIN clinical trial, enzalutamide showed longer time to FACT-P deterioration compared with bicalutamide (median =13.8 vs 8.5 months, p=0.0067).78 On the other hand, in the STRIVE trial, the median time to decline in the enzalutamide patient group was 8.4 months, which was similar to that in the bicalutamide group at 8.3 months (p=0.49).78 The Brief Pain Inventory – Short Form (BPI-SF) questionnaire is a tool to measure pain. In the TERRAIN trial, the mean change of BPI-SF pain score from baseline to week 49 was 0.83 (SD =1.67, 97 patients) in the enzalutamide group and 1.05 (SD =2.00, 48 patients) in the bicalutamide group.78 Taken together, patient satisfaction for enzalutamide was assumed to be equal to or greater than that of bicalutamide. In terms of cost-effectiveness, there are two reports that compare incremental costs between enzalutamide and abiraterone acetate using the data from the PREVAIL and COU-AA-302 studies.79,80 The conclusions were contrary to one another; however, if the adverse event-related cost was taken into consideration, enzalutamide was more reasonable compared with abiraterone acetate.
Conclusion
The AR is the major pathway that controls prostate cancer growth and survival including lethal mCRPC. Targeting the AR indirectly with androgen ablation or directly with antiandrogens has provided decades of therapeutic benefit. A major breakthrough was the clinical development of the antiandrogen enzalutamide for the treatment of mCRPC, and now also used for earlier indications. When looking at the chemical structures of the class of nonsteroidal antiandrogens, there is similarity within all of these compounds through a moiety that binds within the ligand-binding pocket (Figure 1). This similarity in the chemical structure and generally similar mechanisms of action within this class of compounds may provide an opportunity to examine them as a whole and make recommendations to facilitate the development of the next generation of inhibitors that directly bind to the AR. Importantly, the examination of the steady-state Cmin levels (trough levels) reveals that there needs to be sustained plasma levels of antiandrogen in the μg/mL range. At recommended doses for enzalutamide, the steady-state Cmin levels are 11 and 13 μg/mL (for parent compound and active metabolite, respectively) and provide the highest blood levels of all the antiandrogens to date (Figure 2). This is followed by bicalutamide at 9.33 μg/mL, nilutamide between 6 and 7 μg/mL, apalutamide at ~4 μg/mL, darolutamide ranging between 1.8 and 4.3 μg/mL, and flutamide dosed three times a day with 250 mg to achieve a steady-state Cmin of 0.8 μg/mL. If the relative binding affinities to the AR of these antiandrogens are considered, it would be expected, if all other things were equal, that lower blood levels would be permissible for the strong affinity binders such as darolutamide, apalutamide, and enzalutamide, compared with the compounds with less affinity bicalutamide, flutamide, and nilutamide. The fact that enzalutamide has 10× better affinity for the AR compared with bicalutamide would imply that steady-state Cmin levels of bicalutamide are suboptimal for saturation of the AR even though the Cmin is ~9.33 μg/mL. Thus, we strongly encourage examining steady-state Cmin levels at optimal therapeutic dose in preclinical models in order to determine what steady-state Cmin levels are required for clinical efficacy of any newly developed drug that directly binds to the AR, such as has been reported for enzalutamide and apalutamide.53,81 With the clinical development of more novel inhibitors that have unique chemical structures, such as darolutamide, and unique mechanism of action, such as ralaniten acetate, it may be possible to prolong the time to progression and improve the clinical management of mCRPC.
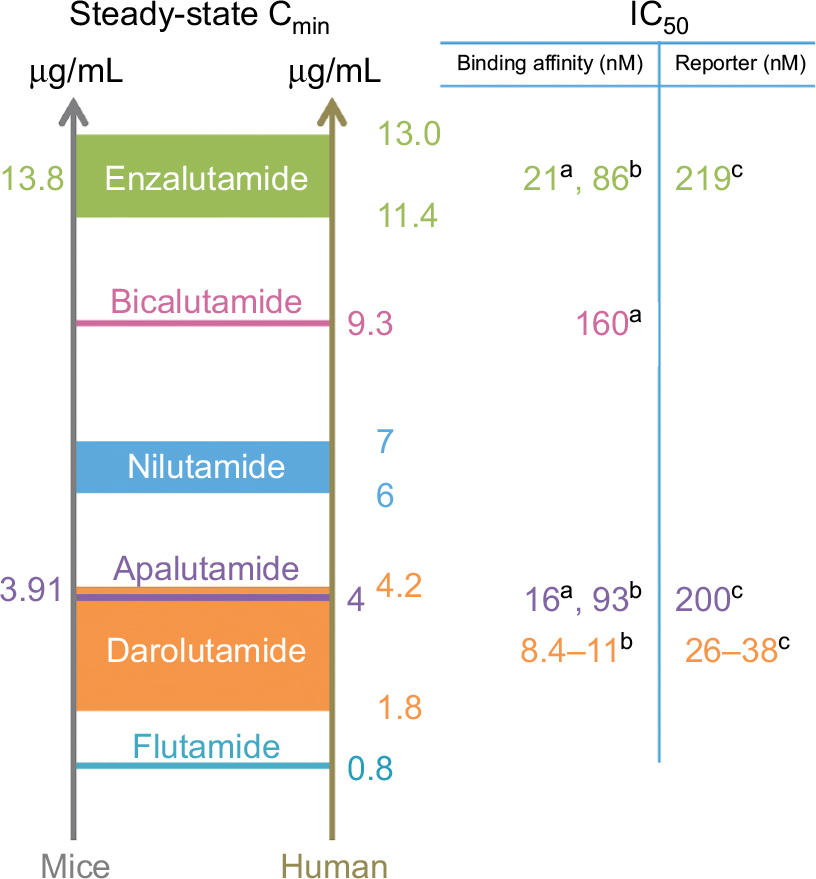 | Figure 2 Steady-state Cmin values with binding affinities and IC50 values for the inhibition of androgen receptor-driven reporter assays. Notes: Values on the left show Cmin levels for optimal therapeutic dose in mice carrying xenografts. Values for humans are from clinical trials with either FDA-approved dose or phase II dose. aBinding affinity measured using 18F-FDHT with LNCaP androgen receptor53 or b[3H]mibolerone with wild-type rat androgen receptor.60 cTranscriptional assay in HEK293 cells.60 Abbreviations: FDA, US Food and Drug Administration; 18F-FDHT, 18F-16b-fluoro-5a-dihydrotestosterone; LNCaP, lymph node carcinoma of the prostate. |
Acknowledgment
This research was supported by a grant to Marianne D Sadar from the US National Cancer Institute, (Grant Number 2R01CA105304).
Author contributions
MDS conceptualized this review. Both YI and MDS did the literature search, reviewed articles, and wrote the manuscript, revised it critically for important intellectual content, approved of the version to be published and agree to be accountable for all aspects of the work
Disclosure
Marianne D Sadar is an inventor of EPI and its analogs including ralaniten acetate (EPI-506) and has licensed the technology to ESSA Pharma. She has shares in ESSA Pharma, is a Director and Officer of ESSA, and receives consulting fees. Yusuke Ito is an inventor. The authors report no other conflicts of interest in this work.
References
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.