")
Back to Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 15
Multi Epitope-Based Vaccine Design for Protection Against Mycobacterium tuberculosis and SARS-CoV-2 Coinfection
Authors Pitaloka DAE, Izzati A, Amirah SR, Syakuran LA
Received 21 March 2022
Accepted for publication 22 July 2022
Published 2 August 2022 Volume 2022:15 Pages 43—57
DOI https://doi.org/10.2147/AABC.S366431
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Alex MacKerell
Dian Ayu Eka Pitaloka,1,2 Afifah Izzati,3 Siti Rafa Amirah,3 Luqman Abdan Syakuran4
1Department of Pharmacology and Clinical Pharmacy, Faculty of Pharmacy, Universitas Padjadjaran, Sumedang, 45363, Indonesia; 2Center of Excellence in Higher Education for Pharmaceutical Care Innovation, Universitas Padjadjaran, Sumedang, 45363, Indonesia; 3Pharmacy Program, Faculty of Pharmacy, Universitas Padjadjaran, Sumedang, 45363, Indonesia; 4Faculty of Biology, Jenderal Soedirman University, Grendeng Purwokerto, 53122, Indonesia
Correspondence: Dian Ayu Eka Pitaloka, Department of Pharmacology and Clinical Pharmacy, Faculty of Pharmacy, Universitas Padjadjaran, Sumedang, 45363, Indonesia, Tel +62-22-84288812, Email [email protected]
Background: A prophylactic and immunotherapeutic vaccine for Mycobacterium tuberculosis (MTB) and SARS-CoV-2 coinfection needs to be developed for a proactive and effective therapeutic approach. Therefore, this study aims to use immunoinformatics to design a multi-epitope vaccine for protection against MTB and SARS-CoV-2 coinfection.
Methods: The bioinformatic techniques were used to screen and construct potential epitopes from outer membrane protein A Rv0899 of MTB and spike glycoprotein of SARS-CoV-2 for B and T cells. The antigenicity, allergenicity, and several physiochemical properties of the developed multi-epitope vaccination were then evaluated. Additionally, molecular docking and normal mode analysis (NMA) were utilized in evaluating the vaccine’s immunogenicity and complex stability.
Results: Selected proteins and predicted epitopes suggest that the vaccine prediction can be helpful in the protection against both SARS-CoV-2 and MTB coinfection. Through docking molecular and NMA, the vaccine-TLR4 protein interaction was predicted to be efficient with a high level of IgG, T-helper cells, T-cytotoxic cells, andIFN-γ.
Conclusion: This epitope-based vaccine is a potentially attractive tool for SARS-CoV-2 and MTB coinfection vaccine development.
Keywords: SARS-CoV-2, Mycobacterium tuberculosis, multi-epitope vaccine, docking simulation
Graphical Abstract:
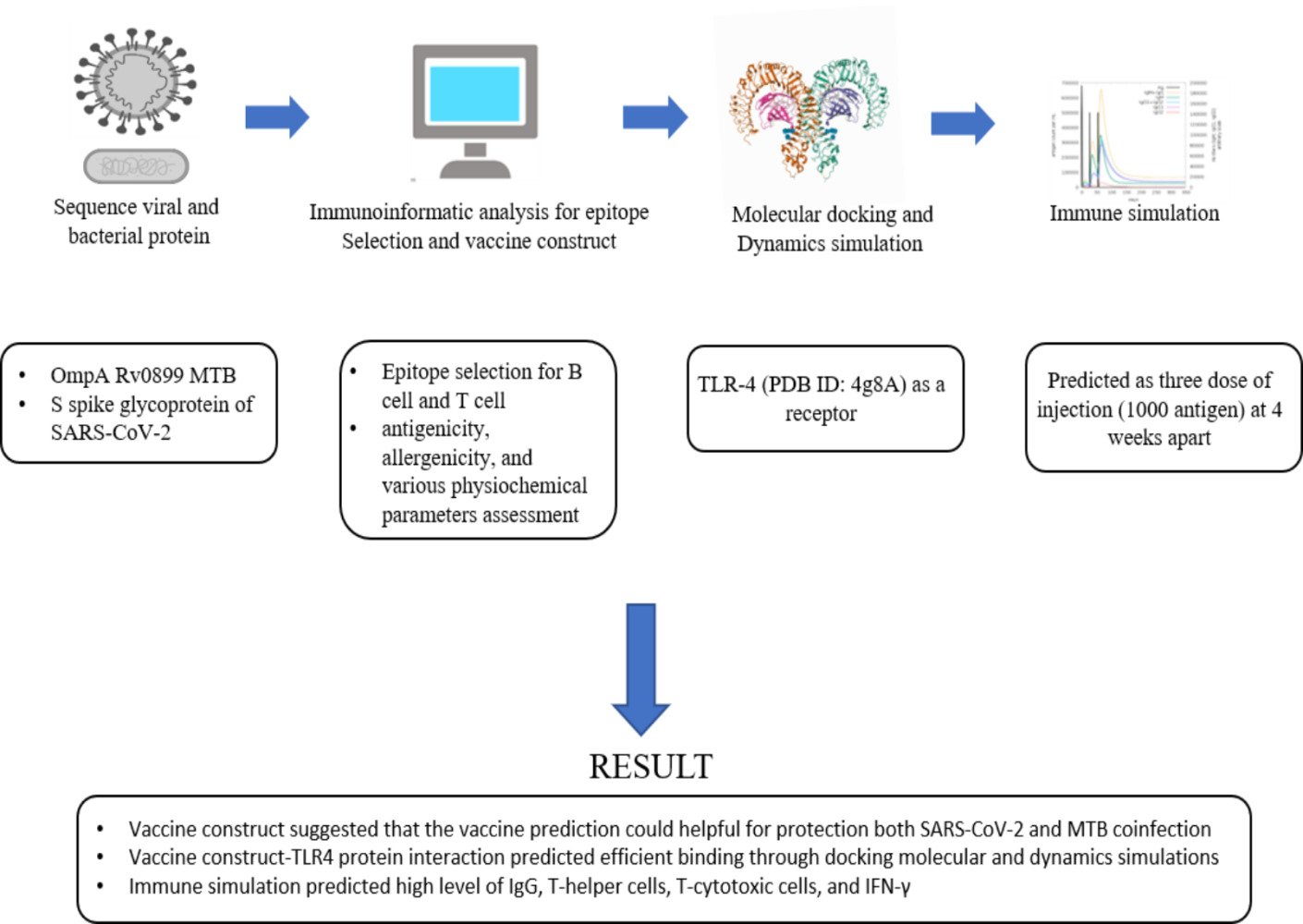
Introduction
The coronavirus disease 2019 (COVID-19) pandemic in numerous regions of the world, including Indonesia, provided a new challenge for global and national efforts in managing infectious diseases. This severe acute respiratory infection appeared in January/February 2020, but the first case was reported in 2019 in China.1,2 While COVID-19 is still very popular in both the scientific literature and the news, other infectious diseases, such as tuberculosis (TB), need to also be considered.3
In Brazil, concomitant infection with Mycobacterium tuberculosis (MTB) and SARS-CoV-2 has been recorded, posing a major health threat.4 Two incidences of coinfection were described in patients with human immunodeficiency virus (HIV). The first case was a 39-year-old patient who was admitted after experiencing fever, myalgia, headache, and cough for seven days. Meanwhile, the second was a 43-year-old male who had a 1-month cough with hemoptoic sputum namely coughing up blood or stained mucus from the bronchi, larynx, trachea, or lungs, that had progressed into mild respiratory distress over the prior seven days. Both individuals had been initially diagnosed with pulmonary TB and contracted SARS-CoV-2 during the 2020 pandemic.4 Based on these findings, serious scientific evaluation of this topic is required.
To eliminate MTB from the human lungs, efficient host defense mechanisms are required, with the bacterial surface playing a crucial role. Membrane stability, molecular transport, and disease are dependent on outer membrane proteins5 such as OmpA, a member of the protein family, which reportedly mediates eukaryotic cell invasion, facilitates serum resistance, and protects against host lung defenses. It also mediates intracellular survival, evasion of host defenses, and stimulation of cytokine production.5 Furthermore, OmpA plays significant pathogenic roles, including adhesion and invasion of bacteria.6 Aside from preventing bacterial infection, inhibiting OmpA can aid in the discovery of novel antimicrobial targets.7
Over the last three years since the COVID-19 pandemic, scientists have investigated the SARS-CoV-2 proteins. The exterior is covered with spike proteins, which are essential for infecting and attacking humans8 by attaching to cells and driving the virus through the cell membrane, allowing cellular invasion.8 The proteolytic activation by the proteases of the host cell is also a crucial determinant.9 However, inducing B-cells and T-cells that can produce immunological responses against the SARS-CoV-2 spike protein offers great potential for combating COVID-19.10
Epitopes are critical for clinical and biological studies because they enable the development of vaccines against diverse and rapidly evolving diseases.11 With the advent of MTB and SARS-Cov-2 coinfections and the associated mortality, innovative and feasible vaccines for preventive protection must be developed continuously. Given that the innate immune system targets outer membrane protein, a reverse vaccinology approach entailing a thorough analysis of the pathogen’s essential characteristics was used to identify immunogenic epitopes against MTB OmpA and SARS-Cov-2 spike glycoprotein which might aid in vaccine development.
Materials and Methods
Sequence Retrieval, Structural, and Physiochemical Analysis
The FASTA sequence of the 326-mer peptidoglycan binding protein OmpA Rv0899 MTB (ID: P9WIU5) and the 1273-mer spike glycoprotein of SARS-CoV-2 (ID: P0DTC2) were obtained from UniprotKB (https://www.uniprot.org/). These proteins, which are abundantly expressed in MTB and SARS-CoV-2 as OmpA and spike glycoprotein respectively, were selected due to their essential role in immune activation and infection of the host.6,9 The GRAVY (Grand average of hydropathicity), half-life, molecular weight, instability index, aliphatic index, and amino acid atomic composition of the protein sequence were bio-computed using the online application Protparam (https://web.expasy.org/protparam/).12
Screening of Potential Epitopes
B Cell Epitopes
The immunogenic epitopes in B-cells were explored using an online server, Bepipred-2.0 (http://tools.iedb.org/bcell/) which uses a hidden Markov model and a propensity scale algorithm to forecast the location of linear B-cell epitopes. This methodology is considered superior to existing sequence-based prediction methods in terms of data collected from solved 3D structures and a massive collection of linear epitopes retrieved from the IEDB database. This server’s epitope threshold was set to 0.5.13
Cytotoxic T Lymphocytes (CTL) Epitopes
The detection of MHC-I antigenic peptides exposed on the target cell surface and recognized by cytotoxic CD8+ T lymphocytes (CTL) is vital for the adaptive immune response.14 The CTL epitopes were predicted with NetCTL.1.2 server (https://services.healthtech.dtu.dk/service.php?NetCTL-1.2) which combines the prediction of peptide MHC class I binding, proteasomal C terminal cleavage, and TAP transport efficiency. Furthermore, the server enables the prediction of CTL epitopes restricted to 12 MHC class I supertypes and has been trained on a collection of 886 known MHC class I ligands, generating accurate prediction results.15 The 9mer-length CTL epitopes were predicted by commonly occurring HLA I alleles, which are estimated to account for >90% of the world’s population. The epitopes with a consensus score of < 2 were considered to be strong binders and were studied further.15
Helper T Lymphocyte (HTL) Epitopes
To identify the HTL epitopes, The Net MHC II pan 3.2 server (https://services.healthtech.dtu.dk/service.php?NetMHCIIpan-3.2) was used with a length of 15m. The server predicts the binding of peptides to MHC class II molecules, which can activate helper CD4+ T lymphocyte (HTL) cells, thereby coordinating and regulating effector cells.14 It also predicts the three human MHC class II isotypes HLA-DR, HLA-DP and HLA-DQ, as well as mouse molecules (H-2). Strong and weak binding peptides are determined based on the percentage Rank calculated by comparing the query peptide’s score to 200,000 random natural peptides of the same length, those with a threshold of 2% were categorized as strong binders.16
Selection of the Epitope Segment
The outcomes of all predictions were compiled and analyzed, then the regions with the highest overlaps were identified. These immunodominant areas were used in subsequent analyses to determine the most suitable epitope domains for vaccine construction. BLASTp (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was performed to ensure that the epitopes did not share homology with humans by identifying similar regions between two sequences.
Selection of Adjuvant and Protein Linker for Vaccine Construction
Adjuvants enhance the efficacy of vaccines by allowing tiny amounts of antigen to induce an effective first immune response and improve memory cell differentiation.17 A 50S ribosomal protein L7/L12 (locus RL7 MYCTU) was selected as an adjuvant (Accession no. P9WHE3) to improve the vaccine’s immunogenicity. The sequence was derived from the UniProt database (https://www.uniprot.org/).18 It was introduced at the vaccine’s N-terminus through an EAAAK linker, while AAY and GPGPG were used to connect the various epitopes. In epitope mapping for the design of multi-epitope vaccines, the AYY, GPGPG, and EAAAK are the most commonly used linkers. They are applied to minimize junctional immunogenicity and boost pathogen-specific immunity.19
Antigenicity, Allergenicity, and Solubility Evaluation
The antigenicity of the vaccine was predicted using ANTIGENpro (http://scratch.proteomics.ics.uci.edu/) and VaxiJen v2.0 (http://www.ddgpharmfac.net/vaxijen/VaxiJen/VaxiJen.html). Antigenpro is a sequence-based, alignment-free, and pathogen-independent antigenicity predictor. It can be used effectively to identify likely protective antigens from an entire proteome.20 Meanwhile, Vaxijen is the first server for alignment-independent prediction of protective antigens based on the auto cross-covariance (ACC) translation of protein sequences into main amino acid vectors.21 The ExPASy Protparam tool (http://web.expasy.org/protparam) was used to evaluate the vaccine candidate’s physicochemical properties.
AllergenFP (http://ddg-pharmfac.net/AllergenFP/) and AllerTOP v.2.0 (http://ddg-pharmfac.net/AllerTOP) were utilized in predicting the vaccine candidate’s non-allergic behavior.22 AllergenFP is a binary classification separating allergens from non-allergens,23 the dataset is characterized by five E-descriptors, and the strings are transformed into uniform vectors using the auto-cross covariance (ACC) transformation.23 Meanwhile, AllerTop is the first alignment-free server for in silico prediction of allergens based on the physicochemical characteristics of proteins.22 It also relies on ACC transformation and E-descriptors.22 The Proso II Protein-sol (http://mbiljj45.bio.med.uni-muenchen.de:8888/prosoII/prosoII.seam) was used to determine the predicted vaccine’s solubility.24 The server is utilized by combining machine-learning-based models and the largest currently available experimental data set provide a substantial advancement in protein solubility predictions.25
Toxicity and Physicochemical Properties Analysis
The toxicity of the constructed vaccine and each of its subunits was predicted with ToxinPred (http://crdd.osdd.net/raghava/toxinpred/) which classifies toxicity and non-toxicity peptides based on an SVM model.26 In particular, the dataset used in this methodology contains 1805 hazardous peptides.26 The constructed vaccine’s physicochemical properties, as well as those of each subunit, were predicted using the ExPASy ProtParam server (https://web.expasy.org/protparam/) which computes various physical and chemical parameters for a given protein sequence, including hydropathicity, charge, half-life, instability index, theoretical isoelectric point value (pI), and molecule weight.27
Secondary Structure Prediction, 3D Homology Modeling, and Refinement of Protein
PSIPRED V3.3 (http://bioinf.cs.ucl.ac.uk/psipred) and RaptorX (http://raptorx.uchicago.edu/%20StructurePropertyPred/predict/) were used to predict alpha-helices, beta-sheets, and coil structures.28,29 It combines multiple feed-forward neural networks that perform analysis on Position Specific Iterated-BLAST (PSI-BLAST) output and also has an average Q3 score of 81.6%, providing a reliable prediction of secondary structure.28 Meanwhile, RaptorX uses a developing machine learning model called DeepCNF (Deep Convolutional Neural Fields) to simultaneously predict secondary structure (SS), solvent accessibility (ACC), and disorder regions (DISO).29
I-TASSER (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) was used to model the three-dimensional structure of the protein target by comparing the similarity index with a template structure available through the protein data bank.30 Refinement was utilized to improve the vaccine candidate’s accuracy using GalaxyRefine (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE).31 Subsequently, the refined structure was validated using RAMPAGE (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php) and the quality of the protein model was determined by plotting the z-score via ProSA-Web (https://prosa.services.came.sbg.ac.at/prosa.php) followed by 3D validation using the PROCHECK server (https://servicesn.mbi.ucla.edu/PROCHECK/).32,33
Molecular Docking of the Vaccine with TLR4
Toll-like receptors (TLRs) are essential sensors of innate immunity that recognize pathogen-associated molecular patterns and initiate signaling for pro-inflammatory cytokine.34 During MTB and SARS-CoV-2 infections, the host’s resistance depends on TLR-4, which rises in human airway epithelial cells,34,35 therefore, understanding the interaction with vaccine candidates is important. The CASTp server (http://sts.bioe.uic.edu/castp/) was used to estimate the binding pockets or cavities in the TLR-4 receptor. It provides identification and measurements of surface-accessible binding pockets as well as information on inner inaccessible cavities for protein molecules.36 Moreover, the multi-epitope vaccination peptide was docked with the TLR4 (PDB ID: 4G8A) receptor using HADDOCK 2.2 (http://haddock.science.uu.nl/services/HADDOCK2.2) to evaluate the ligand-receptor interaction. HADDOCK is a set of Python scripts that calculate structures using crystallography and NMR system.37 Discovery Studio Visualizer (D.S.V.) v16.1.0.153500 was used to visualize the docked model.37–39
Normal Mode Analysis of the Vaccine-Receptor Complex
The simulation of a protein’s stability was carried out with the iMOD server (iMODS) (http://imods.chaconlab.org) using Normal Mode Analysis (NMA) to compute its internal coordinates (NMA). The stability of the protein was shown by the main-chain deformability plot, the B-factor values, the eigenvalue, the covariance matrix, and the elastic network model.40
In silico Cloning Optimization of Designed Vaccine Construct
Codon optimization is a critical aspect for successful protein expression, particularly when a heterologous expression method is applied.41 To express the vaccine candidate, the E. coli strain K12 was selected as the host organism. It is classified as nonpathogenic to humans due to the outer membrane’s dysfunctional LPS core, which prevents adhesion to gut mucosa, and its inability to express the capsular antigens required for colonization and virulence.42 Due to the difference in codon usage between humans and E. coli, the Java Codon Adaptation Tool (JCat) server (http://www.prodoric.de/JCat) was used to adapt the codons to the prokaryotic organism, thereby improving the expression rate.
Immune Simulation
In silico immunological simulations were performed using the C-ImmSim server (http://150.146.2.1/C-IMMSIM/index.php),43 which is an innovative method for analyzing the immune systems of mammals. The program provides a mesoscopic scale immune system simulator with machine learning techniques for molecular-level predictions of major histocompatibility complex (MHC)–peptide-binding interactions, linear B-cell epitope finding, and protein–protein potential estimation.43 In this study, all simulation parameters were kept at their default parameters, and three dose injections of 1000 antigens were administered four weeks apart, then each response was examined.
Results
Epitope Prediction
B-Cell Epitope Prediction
B-cells play a primary role in humoral immunity, therefore, a specific epitope for their receptors is critical for vaccine production to generate antibodies The BepiPred 2.0 server was used to predict linear B-cell epitopes of varying lengths. It predicted 2 epitopes for OmpA Rv0899 MTB from 6 epitopes and 4 for SARS-CoV-2 spike glycoprotein from 8 epitopes. A total of 6 B-cell epitopes were selected based on various criteria, such as antigenicity and similarity. The selected epitopes then finalized to be further screened for the vaccine candidate (Table 1).
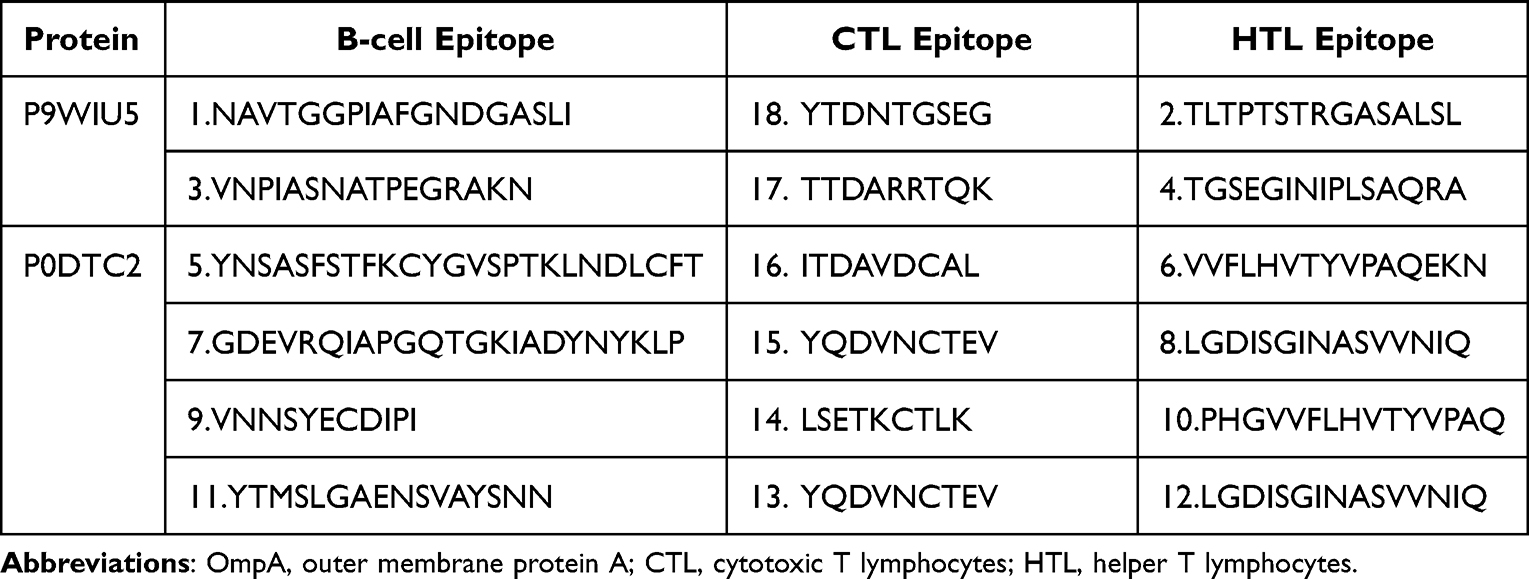 |
Table 1 Predicted B-Cell and T-Cell Epitopes Selected to Design the Multi-Epitope Peptide-Based Vaccine. Serial Numbers Attached to Epitopes Show Their Order of Appearance in the Vaccine’s Design |
Cytotoxic T Lymphocyte (CTL) Prediction
A total of 273 CTL ligands for OmpA of MTB were identified using the NetCTL 1.2 server. Out of 273, total of 5 CTL (9-mer) ligands were selected with the default epitope identification cutoff score for OmpA Rv0899 MTB set to 0. Additionally, 4 were selected for further screening among 37 predicted CTL ligands for the spike glycoprotein of SARS-CoV-2. Nine epitopes in total were selected due to their high ratings and were used to generate the vaccine candidate (Table 1).
Helper T Lymphocyte (HTL) Prediction
HTL epitopes were identified as highly binding MHC-II epitopes for the human alleles HLA-DR, HLA-DQ, and HLA-DP which were predicted using the NetMHCII 2.2 web server based on their IC50 values. Two epitopes (15-mer) were selected for OmpA Rv0899 MTB from 64 high-binding HTL, and four (15-mer) were used for SARS-CoV-2 spike glycoprotein from 53. Six HTL epitopes in total were used for the construction of the vaccine candidate (Table 1).
Construction of a Multi-Epitope Vaccine
The vaccine candidate was designed using a total of six B-cell epitopes, nine CTL and six HTL, then AAY and GPGPG linkers were employed to join predicted peptide sequences comprising B- and T-cell epitopes. The 50S ribosomal L7/L12 (Locus RL7 MYCTU) was selected as an adjuvant and linked to the amino terminus of the vaccine peptide through an EAAAK linker to enhance antigen-specific immune responses. Additionally, a C-terminal 6xHis tag was introduced to aid in protein purification and identification. The final vaccine contained 472 amino acid residues drawn from 21 peptide sequences that had been combined (Figure 1).
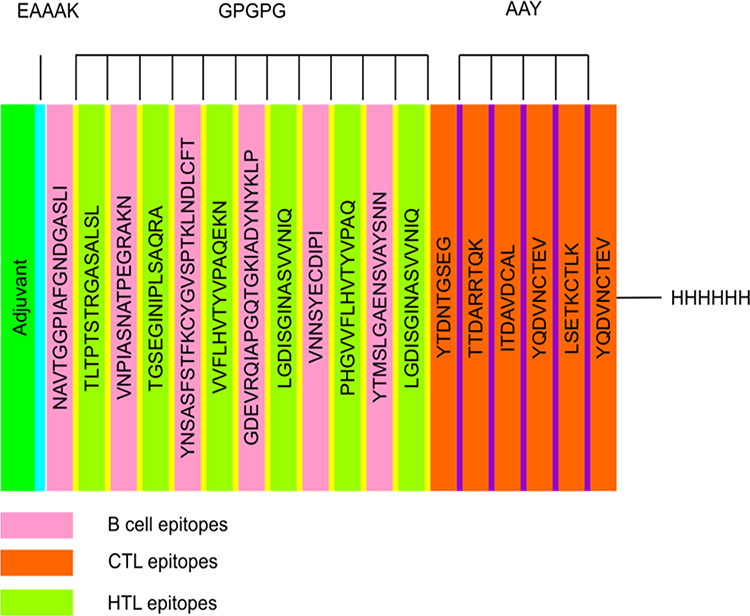 |
Figure 1 Schematic presentation of the multi-epitope vaccine. The 472-amino acid long peptide sequence contains an adjuvant (green) at the amino-terminal end and is connected to the multi-epitope sequence via an EAAAK linker (cyan). B and HTL epitopes are linked using GPGPG linkers (yellow), whereas CTL epitopes (Orange) are linked using AAY linkers (purple). At the Carboxy terminus, a 6x-His tag is inserted for purification and identification purposes. |
Prediction of the Antigenicity and Allergenicity of the Vaccine Construct
The VaxiJen 2.0 server projected the constructed vaccine’s antigenicity to be 0.8211 for bacteria and 0.5472 for viruses at a threshold of 0.5 and 0.9319 for ANTIGENpro. These results show that both of the produced sequences are naturally antigenic. Additionally, both the AllerTOP v.2 and AllergenFP servers demonstrated that the constructed vaccine is non-allergenic.
Physiochemical Properties and Solubility Prediction of the Vaccine Construct
The molecular weight of the constructed vaccine was predicted to be 47.75 kDA with a theoretical isoelectric point value (pI) of 4.97. Based on the pI value, it was expected that the protein will be acidic in nature. The half-life was 30 hours in vitro in human reticulocytes, > 20 hours in-vivo in yeast, and > 10 hours in-vivo in E. coli. Additionally, with a predicted solubility score of 0.62, the protein was expected to be soluble upon expression. The instability index (II) was 21.87, which is considered stable, while the aliphatic index was 76.99. GRAVY predicted a score of −0.153, indicating that the molecule is hydrophilic.
Secondary Structure Prediction
The constructed vaccine was estimated to comprise 21% alpha-helix, 15% beta-strand, and 62% coil as shown in Figure 2. Additionally, 56% of amino acid residues were expected to be exposed to solvent, 22% to medium, and 21% hidden. The RaptorX Property server showed that 62 residues (13%) were placed in disordered domains.
 |
Figure 2 A graphical representation of secondary structure features of the constructed vaccine sequence. The protein was predicted to comprise alpha-helices (21%), beta strands (15%), and coils (62%). |
Tertiary Structure Modelling
A 3D structure model of the vaccine was created using the I-TASSER server which predicted five tertiary models for the proposed protein using ten template sequences, including 2D4Y, 3VA7, 4UT1, 5M59, 5XBJ, 6UEB, 6W2J, 5D06, 6XJA, and 7CPX. According to their Z-score values, each was well aligned ranging from 1.18 to 5.46. C-score values for the five projected models ranged from 4.56 to 0.83. Typically, the C-score ranges between 5 and 2, with higher values suggesting greater confidence.30 The model with the highest value from the homology modeling was selected for further refinement (Figure 3A). The selected model was estimated to have a TM-score of 0.61±0.14 and a Root Mean Square Deviation (RMSD) of 9.14.6. Meanwhile, the TM-score has been developed as a scale for comparing the similarity between two structures.44 It was designed to circumvent the RMSD’s sensitivity to local inaccuracy, a value greater than 0.5 implies a model with correct topology, while values less than 0.17 indicate random similarity.45 These cut-off values are independent of the protein length.
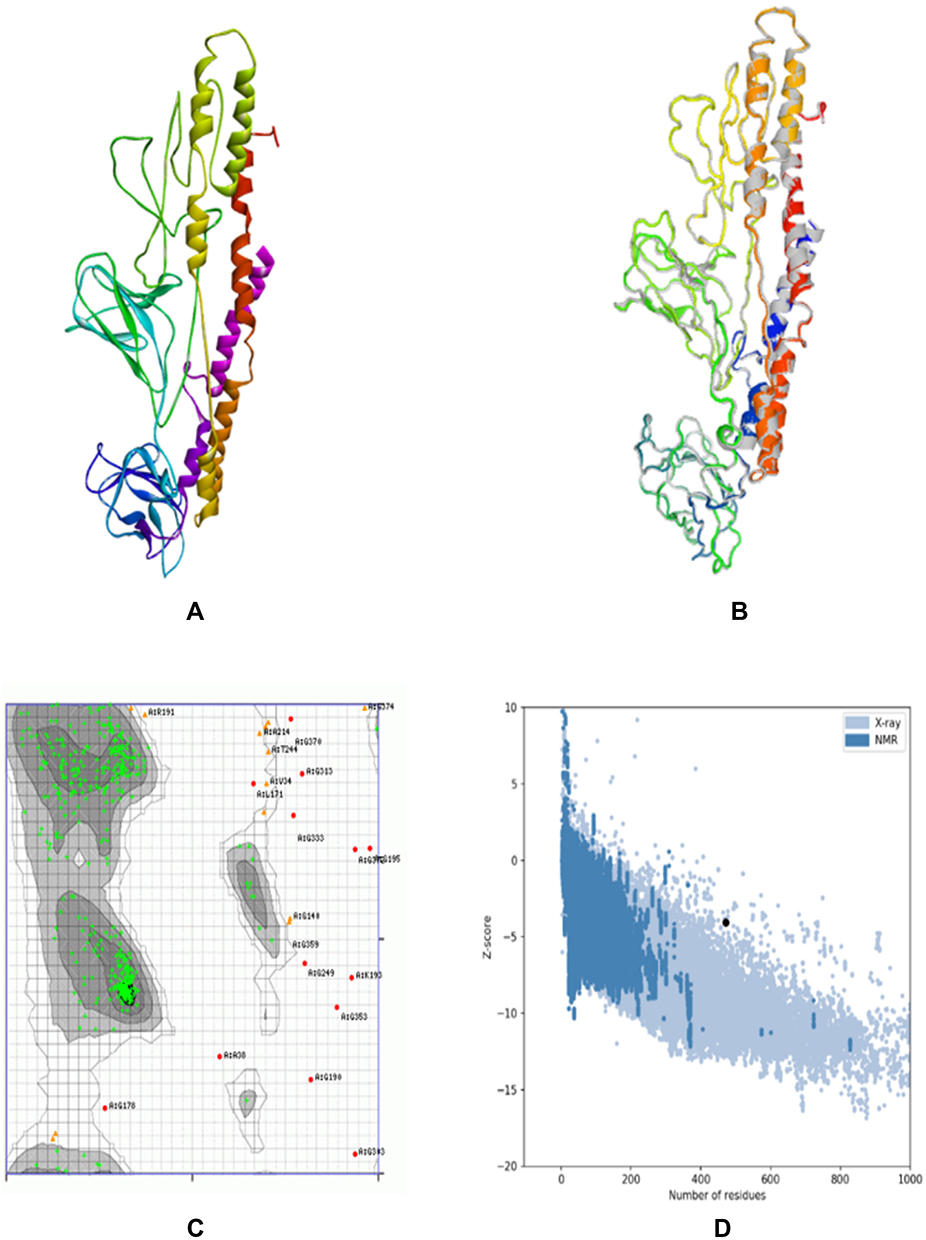 |
Figure 3 Protein modeling, refinement, and validation for constructed vaccine. The final 3D model of the multi-epitope vaccine was obtained after homology modeling on I-TASSER (A). Superimposed of the refined 3D structure (colored) on the “crude model” (grey) by GalaxyRefine (B). Validation of the refined model with Ramachandran plot analysis showed 94.47%, 2.77%, and 2.76% of protein residues in favored, allowed, and disallowed (outlier) regions, respectively (C), and ProSA-web gave a Z-score of −4.07 (D). |
Tertiary Structure Refinement
The final vaccination was refined on ModRefiner and then on the GalaxyRefine server, to yield five models. Model 2 was selected as the best based on a variety of metrics, including GDT-HA 0.9454, RMSD 0.432, and MolProbity 2.34. The clash score was 16.6, the poor rotamers rating was 1.2, and the Ramachandran plot score was 89.1%. Therefore, this model was selected as the vaccine candidate for analysis (Figure 3B).
Tertiary Structure Validation
The Ramachandran plot analysis of the modeled protein revealed that 94.47% of residues are in preferential regions, this is consistent with the 94.5% score predicted by the GalaxyRefine analysis. Additionally, 2.77% and 2.76% of the residues were in the allowed and disallowed regions respectively as shown in Figure 3C. ProSA-web was used to verify the quality and possibility for error in the crude 3D model. After refinement, the selected model had an overall Z-score of 4.07 with ProSA-web for the input vaccine protein model (Figure 3D).
Molecular Docking of the Final Vaccine Construct with TLR4
The CASTp server was used to determine the protein binding and hydrophobic interaction sites on the surface. A binding pocket was identified based on a previous study with the same PDB ID46 which found that the TLR4 binding pocket between residues 1 and 298 might serve as a potential binding site. The pocket and mouth had a molecular surface area of 325.12 Å2 and 197.82 Å respectively with a molecular surface volume of 581.33 Å and the molecular circumference total was 80 Å.47 Furthermore, the immune response of TLR-4 against multi-epitope vaccine was estimated by analyzing the overall conformational stability of the vaccine-TLR4 docked complex. The following active interface amino acid residues were found; P23, S25, T26, P28, V32, V33, I36, G39, M41, G42, L43, A44, P45, T46, L47, I48, A50, A51, P53, P54, S55, L57, A58, A60, P63, A67, H68, G70, S71, T72, S73, P75, S76, P78, G79, A87, T92, A95, G96, G99, S100, S102, S115, L119, G124, G523, H529, A544, G547, S569, S570, T626, and C627 from the B chain of TLR4 (Figure 4). The top-ranking pose for each docked complex with the lowest total intermolecular energies was 2128.649 Kcal/mol.
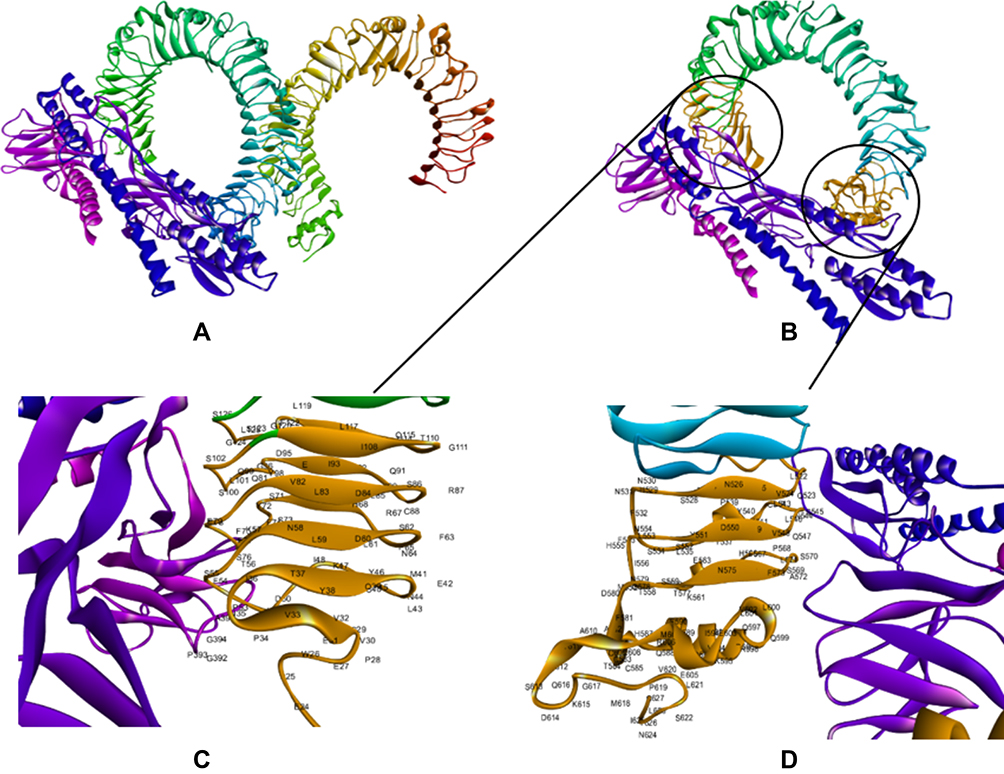 |
Figure 4 Docking of constructed vaccine with the immune receptor (TLR4). Docked for the final vaccine-TLR4 complex (A). Interaction of multi-epitope vaccine with B chain of TLR4 is highlighted in Orange (B). The interacting residues of the multi-epitope vaccine and B chain of TLR 4 (C and D). The docked model was visualized using Discovery Studio Visualizer (D.S.V.) v16.1.0.153500. |
Normal Mode Analysis of the Vaccine-Receptor Complex
NMA were carried out by the iMOD server to determine the stability and physical mobility of the vaccine-TLR4 docked complex.48 The deformability of the main-chain is shown in Figure 5A, while hinged locations were visualized using the high strain. The B-factor values calculated using NMA were proportional to RMSD value (Figure 5B). Furthermore, the eigenvalues of the complex numbers in Figure 5C were 4.344 x 10−5, which was closely connected to the energy necessary to deform the structure. Correlations were also detected in the covariance matrix between pairs of residues (Figure 5D). The elastic network model assumed that there was a connection between the atom and the spring (Figure 5E). NMA confirmed the stability of the vaccine candidate model.
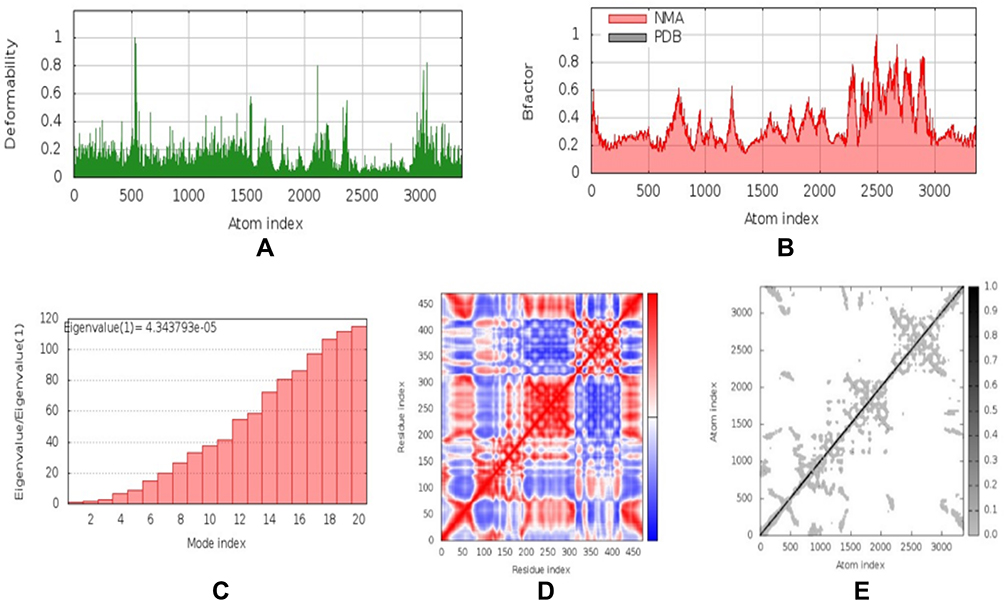 |
Figure 5 Normal mode analysis (NMA) modeling of docked vaccine-TLR4 complexes. Modeling the deformability of the main chain, the hinge is an area with high deformability (A). B-factor values were calculated from NMA to quantify the uncertainty of each atom (B). Eigenvalues of adjacent complexes represent the energy required to deform the structure (C). Covariance matrix between pairs of residues (red: correlated, white: uncorrelated, blue: anti-correlated) (D). Elastic network model assuming bonding between atoms and springs; the darker the grey, the stiffer the spring (E). |
Codon Optimization of the Final Vaccine Construct
The Java Codon Adaptation Tool (JCat) was used to optimize codon usage of the vaccine candidate in E. coli (strain K12) for maximal protein expression. The optimized codon sequence was 165 nucleotides in length, while the enhanced nucleotide has a Codon Adaptation Index (CAI) of 1.0 with an average GC content of 39.40%, indicating the possibility of the vaccine candidate expression in the E. coli host, because the ideal range of GC content is between 30% and 70%.
Immune Simulation
The immunological responses were simulated with C-ImmSim and the result showed that there was an increase in the IgM + IgG antibodies (Figure 6A). This pattern implies the establishment of immunological memory, and also greater antigen clearance following subsequent exposures. A similarly high response was observed in the TH (helper) and TC (cytotoxic) cell populations with corresponding memory development (Figure 6B and C). Repeated exposure with 12 injections four-week apart demonstrated that the IFN-γ and TH cell population remained at high levels throughout the duration of exposure (Figure 6D).
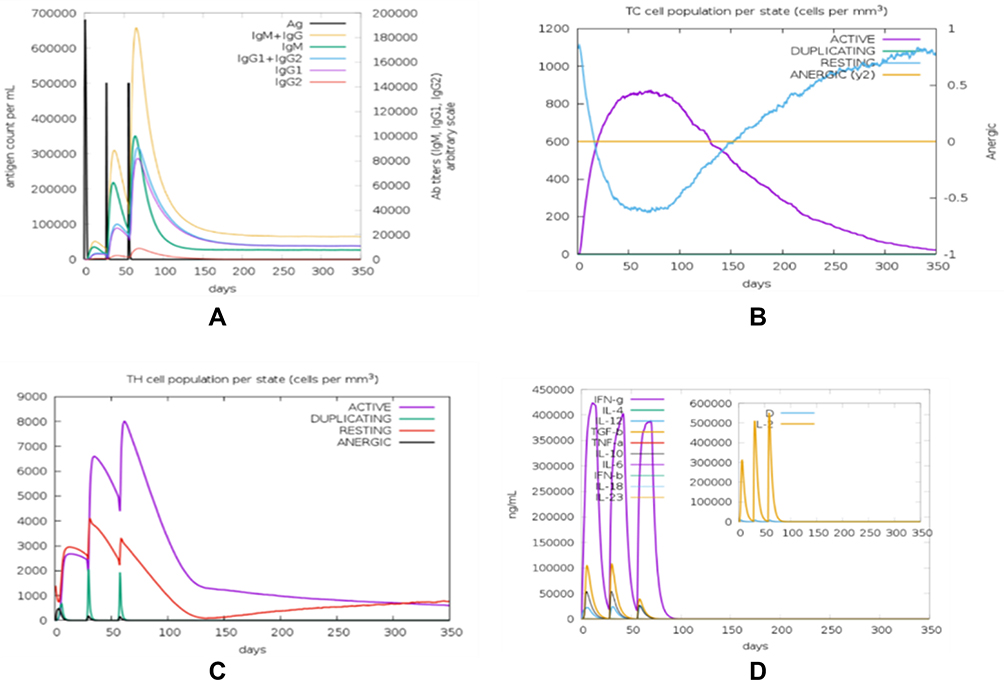 |
Figure 6 Result of immune response simulation revealed that the final vaccine has the potential to induce antibody-mediated humoral immunity (A), CD8+ T Cell immune response (B), CD4+ T cell immune response (C) and non-specific immune response mediated by cytokines production (D). |
Discussion
The advent of a new SARS-CoV-2 strain is a global threat that has claimed the lives of a large number of individuals worldwide, including healthcare workers. It is transferred from one person to another through airborne droplets.49 Vaccination regimens against this virus have been created successfully, leading to an improvement in global human health statistics.50 However, co-infection with MTB is regarded as a key risk factor for the severity as well as mortality rates, and the scientific basis for this issue is unknown.51 Immunoinformatics methods have changed the scope of vaccines’ preparation and might provide a solution to prevent this problem.
The vaccine candidate has a molecular weight of 47.75 kDa and a predicted soluble weight of 0.62 following overexpression. The solubility in the overexpressed state was satisfactory and is a necessary condition for various biochemical and functional studies.52 Furthermore, the protein is predicted to be acidic with a theoretical PI of 4.97 and stable with a score of 21.87 on the instability index. A protein with an instability index of less than 40 is stable, while those with more than 40 are almost certainly unstable.53 The vaccine design had a predicted aliphatic index of 76.99, which shows that the side chains were aliphatic, indicating a high potential for thermostability. The aliphatic index information is critical for proteins with a molecular weight of less than 100,000.54 The proposed vaccine is expected to be non-allergenic with an antigen prediction value greater than the threshold, also, it is classified as an antigen and might be subjected to additional examination.
The immunoinformatics approach was used to construct a multi-epitope vaccine that comprises B-cell, TH cell, and TC cell epitopes from spike glycoprotein of SARS-CoV-2 and OmpA of MTB. This method has several advantages over the whole protein-based vaccine, for example, it can stimulate immune system responses to protective epitopes without causing an autoimmune reaction.55 Also, given that the vaccine only has expected immunodominant epitopes, it is less likely to react with non-immunodominant or non-neutralizing types.56
Non-neutralizing epitopes potentially contribute to the antibody-dependent enhancement (ADE) observed during dengue virus infection.57 Adding a genetically fused protein with adjuvant activity such as The 50S ribosomal L7/L12 will make the vaccine more effective and prolong the immune response.58 Given the benefits of vaccination based on several epitopes, this method was employed to produce M-001, a multi-epitope-based universal influenza vaccine. Clinical tests reported that it strongly boosts both cellular and humoral immune responses.59 Moreover, an in silico multi-epitope tetravalent dengue vaccine which has been shown to elicit significant levels of IFN- and neutralizing antibodies has also been developed.60
Multi-epitope vaccines containing B-cell and T-cell epitopes can induce both humoral and cellular immunity, thereby preventing undesirable side effects associated with the additional epitope produced in response to intact protein antigens. This is important because undesired immune responses have the potential to induce an autoimmune response. Additionally, it has an effect on both specific and protective immune responses.61 The immune response simulation demonstrated that the developed vaccine was capable of inducing humoral, CD4+ T cell, and CD8+ T cell immunity.
Most preventative vaccines against infectious pathogens have focused on making neutralizing antibodies. However, a few studies showed that neutralizing antibodies alone are not enough to give long-lasting immunity and protection against certain pathogens, hence, T cell immunity is also needed.62 IFN production by CD8+ and CD4+ is essential for MTB control,63 while T cells also aid in viral infection control by releasing cytokines such as IFN- and TNF. A recent study demonstrated that the specific memory CD4+ and CD8+ T cell response to SARS-CoV-2 is relatively long-lasting compared to neutralizing antibodies.64 Also, it is more difficult for a virus to hide from memory T cell responses caused by the SARS-CoV-2 vaccine or by a natural infection.65
Limitation
This study did not provide the cross-protection results expected because the vaccine was engineered from each part of the respected pathogen, which has different abilities.
Conclusion
The in silico approach is the first step towards designing a vaccine for achieving control of SARS-CoV-2 and MTB coinfection. In this study, immunoinformatic tools were used to design a potential vaccine peptide coding in multiple B-cell and T-cell epitopes for these two pathogens. The spike protein of SARS-CoV-2 and outer membrane A of MTB were used due to their essential role in immune activation and infection of the host immune system. The results showed that the vaccine peptide designed can prevent SARS-CoV-2 and MTB coinfection, as well as help in boosting the host immune system. Further investigations are needed to determine the vaccine’s efficacy using laboratory and animal models.
Acknowledgments
This study was supported by Directorate of Research and Community Service Universitas Padjadjaran.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395(10223):507–513. doi:10.1016/S0140-6736(20)30211-7
2. Wu Z, McGoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA. 2020;323(13):1239–1242. doi:10.1001/jama.2020.2648
3. Ong CWM, Goletti D. Impact of the global COVID-19 outbreak on the management of other communicable diseases. Int J Tuberc Lung Dis. 2020;24(5):547–548. doi:10.5588/ijtld.20.0140
4. Gadelha Farias LAB, Gomes Moreira AL, Austregésilo Corrêa E, et al. Case Report: coronavirus Disease and Pulmonary Tuberculosis in Patients with Human Immunodeficiency Virus: report of Two Cases. Am J Trop Med Hyg. 2020;103(4):1593–1596. doi:10.4269/ajtmh.20-0737
5. Danilchanka O, Sun J, Pavlenok M, et al. An outer membrane channel protein of Mycobacterium tuberculosis with exotoxin activity – PubMed. National Acad Sci. 2014;111(18):6750. doi:10.1073/pnas.1400136111
6. Marassi FM. Mycobacterium tuberculosis Rv0899 defines a family of membrane proteins widespread in nitrogen-fixing bacteria. Proteins. 2011;79(10):2946–2955. doi:10.1002/prot.23151
7. Sundar S, Thangamani L, Piramanayagam S. Computational identification of significant immunogenic epitopes of the putative outer membrane proteins from Mycobacterium tuberculosis. J Genet Eng Biotechnol. 2021;19(1):48. doi:10.1186/s43141-021-00148-9
8. Lan J, Ge J, Yu J, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–220. doi:10.1038/s41586-020-2180-5
9. Takeda M. Proteolytic activation of SARS‐CoV‐2 spike protein. Microbiol Immunol. 2022;66(1):15–23. doi:10.1111/1348-0421.12945
10. Sternberg A, Naujokat C. Structural features of coronavirus SARS-CoV-2 spike protein: targets for vaccination. Life Sci. 2020;257:118056. doi:10.1016/j.lfs.2020.118056
11. Soria-Guerra RE, Nieto-Gomez R, Govea-Alonso DO, Rosales-Mendoza S. An overview of bioinformatics tools for epitope prediction: implications on vaccine development. J Biomed Inform. 2015;53:405–414. doi:10.1016/j.jbi.2014.11.003
12. Wilkins MR, Gasteiger E, Bairoch A, et al. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol Clifton NJ. 1999;112:531–552. doi:10.1385/1-59259-584-7:
13. Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017;45(W1):W24–W29. doi:10.1093/nar/gkx346
14. Wieczorek M, Abualrous ET, Sticht J, et al. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: conformational Plasticity in Antigen Presentation. Front Immunol. 2017;8:292. doi:10.3389/fimmu.2017.00292
15. Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007;8:424. doi:10.1186/1471-2105-8-424
16. Jensen KK, Andreatta M, Marcatili P, et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology. 2018;154(3):394–406. doi:10.1111/imm.12889
17. Bastola R, Noh G, Keum T, et al. Vaccine adjuvants: smart components to boost the immune system. Arch Pharm Res. 2017;40(11):1238–1248. doi:10.1007/s12272-017-0969-z
18. Chauhan V, Rungta T, Goyal K, Singh MP. Designing a multi-epitope based vaccine to combat Kaposi Sarcoma utilizing immunoinformatics approach. Sci Rep. 2019;9(1):2517. doi:10.1038/s41598-019-39299-8
19. Ayyagari VS, Venkateswarulu TC, Abraham PK, Srirama K. Design of a multi-epitope-based vaccine targeting M-protein of SARS-CoV2: an immunoinformatics approach. J Biomol Struct Dyn. 2022;40(7):2963–2977. doi:10.1080/07391102.2020
20. Magnan CN, Zeller M, Kayala MA, et al. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics. 2010;26(23):2936–2943. doi:10.1093/bioinformatics/btq551
21. Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007;8:4. doi:10.1186/1471-2105-8-4
22. Dimitrov I, Bangov I, Flower DR, Doytchinova I. AllerTOP v.2–a server for in silico prediction of allergens. J Mol Model. 2014;20(6):2278. doi:10.1007/s00894-014-2278-5
23. Dimitrov I, Naneva L, Doytchinova I, Bangov I. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinforma Oxf Engl. 2014;30(6):846–851. doi:10.1093/bioinformatics/btt619
24. Hebditch M, Carballo-Amador MA, Charonis S, Curtis R, Warwicker J. Protein–Sol: a web tool for predicting protein solubility from sequence. Bioinformatics. 2017;33(19):3098–3100. doi:10.1093/bioinformatics/btx345
25. Smialowski P, Doose G, Torkler P, Kaufmann S, Frishman D. PROSO II–a new method for protein solubility prediction. FEBS J. 2012;279(12):2192–2200. doi:10.1111/j.1742-4658.2012.08603.x
26. Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS One. 2013;8(9):e73957. doi:10.1371/journal.pone.0073957
27. Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31(13):3784–3788. doi:10.1093/nar/gkg563
28. Buchan DWA, Jones DT. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019;47(W1):W402–W407. doi:10.1093/nar/gkz297
29. Kallberg M, Wang H, Wang S, et al. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7(8):1511–1522. doi:10.1038/nprot.2012.085
30. Zheng W, Zhang C, Bell EW, Zhang Y. I-TASSER server for protein structure and function prediction. Future Gener Comput Syst. 2019;99:73–85. doi:10.1093/nar/gkv342
31. Heo L, Park H, Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013;41:384–388. doi:10.1093/nar/gkt458
32. Laskowski RA, MacArthur MW, Thornton J. PROCHECK: validation of protein structure coordinates. In: Rossmann MG, Arnold E, editors. Nternational Tables of Crystallography, Volume F. Crystallography of Biological Macromolecules. The Netherlands: Dordrecht, Kluwer Academic Publishers; 2001:722–725.
33. Lovell SC, Davis IW, Arendall WB, et al. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50(3):437–450. doi:10.1002/prot.10286
34. Kayesh MEH, Kohara M, Tsukiyama-Kohara K. An Overview of Recent Insights into the Response of TLR to SARS-CoV-2 Infection and the Potential of TLR Agonists as SARS-CoV-2 Vaccine Adjuvants. Viruses. 2021;13(11):2302. doi:10.3390/v13112302
35. Sepehri Z, Kiani Z, Kohan F, Ghavami S. Toll-Like Receptor 4 as an Immune Receptor Against Mycobacterium tuberculosis: a Systematic Review. Lab Med. 2019;50(2):117–129. doi:10.1093/labmed/lmy047
36. Binkowski TA, Naghibzadeh S, Liang J. CASTp: computed Atlas of Surface Topography of proteins. Nucleic Acids Res. 2003;31(13):3352–3355. doi:10.1093/nar/gkg512
37. Dominguez C, Boelens R, Bonvin AMJJ. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125(7):1731–1737. doi:10.1021/ja026939x
38. Xue LC, Rodrigues JP, Kastritis PL, Bonvin AM, Vangone A. PRODIGY: a web server for predicting the binding affinity of protein-protein complexes. Bioinforma Oxf Engl. 2016;32(23):3676–3678. doi:10.1093/bioinformatics/btw514
39. de Vries SJ, Bonvin AMJJ. CPORT: a consensus interface predictor and its performance in prediction-driven docking with HADDOCK. PLoS One. 2011;6(3):e17695. doi:10.1371/journal.pone.0017695
40. Lopez-Blanco JR, Aliaga JI, Quintana-Orti ES, Chacon P. iMODS: internal coordinates normal mode analysis server. Nucleic Acids Res. 2014;42:W271–6. doi:10.1093/nar/gku339
41. Al-Hawash AB, Zhang X, Ma F. Strategies of codon optimization for high-level heterologous protein expression in microbial expression systems. Gene Rep. 2017;9:46–53. doi:10.1016/j.genrep.2017.08.006
42. Blattner FR, Plunkett G, Bloch CA, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277(5331):1453–1462. doi:10.1126/science.277.5331.1453
43. Rapin N, Lund O, Bernaschi M, Castiglione F. Computational Immunology Meets Bioinformatics: the Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS One. 2010;5(4):e9862. doi:10.1371/journal.pone.000986244
44. Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins. 2004;57(4):702–710. doi:10.1002/prot.20264
45. Xu J, Zhang Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinforma Oxf Engl. 2010;26(7):889–895. doi:10.1093/bioinformatics/btq066
46. Devi A, Chaitanya NSN. In silico designing of multi-epitope vaccine construct against human coronavirus infections. J Biomol Struct Dyn. 2021;39(18):6903–6917. doi:10.1080/07391102.2020
47. Shey RA, Ghogomu SM, Esoh KK, et al. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci Rep. 2019;9(1):4409. doi:10.1038/s41598-019-40833-x
48. Yang Z, Bogdan P, Nazarian S. An in silico deep learning approach to multi-epitope vaccine design: a SARS-CoV-2 case study. Sci Rep. 2021;11(1):3238. doi:10.1038/s41598-021-81749-9
49. Jayaweera M, Perera H, Gunawardana B, Manatunge J. Transmission of COVID-19 virus by droplets and aerosols: a critical review on the unresolved dichotomy. Environ Res. 2020;188:109819. doi:10.1016/j.envres.2020.109819
50. Schoch-Spana M, Brunson EK, Long R, et al. The public’s role in COVID-19 vaccination: human-centered recommendations to enhance pandemic vaccine awareness, access, and acceptance in the United States. Vaccine. 2021;39(40):6004–6012. doi:10.1016/j.vaccine.2020.10.059
51. Mirzaei R, Goodarzi P, Asadi M, et al. Bacterial co-infections with SARS-CoV-2. IUBMB Life. 2020;72(10):2097–2111. doi:10.1002/iub.2356
52. Khatoon N, Pandey RK, Prajapati VK. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci Rep. 2017;7(1):8285. doi:10.1038/s41598-017-08842-w
53. Kaur A, Pati PK, Pati AM, Nagpal AK. Physico-chemical characterization and topological analysis of pathogenesis-related proteins from Arabidopsis thaliana and Oryza sativa using in-silico approaches. PLoS One. 2020;15(9):e0239836. doi:10.1371/journal.pone.0239836
54. Nehete JY, Bhambar RS, Narkhede MR, Gawali SR. Natural proteins: sources, isolation, characterization and applications. Pharmacogn Rev. 2013;7(14):107–116. doi:10.4103/0973-7847.120508
55. Meza B, Ascencio F, Sierra-Beltrán AP, Torres J, Angulo C. A novel design of a multi-antigenic, multistage and multi-epitope vaccine against Helicobacter pylori: an in silico approach. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis. 2017;49:309–317. doi:10.1016/j.meegid.2017.02.007
56. Murugavelu P, Perween R, Shrivastava T, et al. Non-neutralizing SARS CoV-2 directed polyclonal antibodies demonstrate cross-reactivity with the HA glycans of influenza virus. Int Immunopharmacol. 2021;99:108020. doi:10.1016/j.intimp.2021.108020
57. Rammensee HG, Gouttefangeas C, Heidu S, et al. Designing a SARS-CoV-2 T-Cell-Inducing Vaccine for High-Risk Patient Groups. Vaccines. 2021;9(5):428. doi:10.3390/vaccines9050428
58. Zhang L. Multi-epitope vaccines: a promising strategy against tumors and viral infections. Cell Mol Immunol. 2018;15(2):182–184. doi:10.1038/cmi.2017.92
59. Romeli S, Hassan SS, Yap WB. Multi-Epitope Peptide-Based and Vaccinia-Based Universal Influenza Vaccine Candidates Subjected to Clinical Trials. Malays J Med Sci. 2020;27(2):10–20. doi:10.21315/mjms2020.27.2.2
60. Chan Y, Jazayeri SD, Ramanathan B, Poh CL. Enhancement of Tetravalent Immune Responses to Highly Conserved Epitopes of a Dengue Peptide Vaccine Conjugated to Polystyrene Nanoparticles. Vaccines. 2020;8(3):E417. doi:10.3390/vaccines8030417
61. Lim HX, Lim J, Jazayeri SD, Poppema S, Poh CL. Development of multi-epitope peptide-based vaccines against SARS-CoV-2. Biomed J. 2021;44(1):18–30. doi:10.1016/j.bj.2020.09.005
62. Panagioti E, Klenerman P, Lee LN, van der Burg SH, Arens R. Features of Effective T Cell-Inducing Vaccines against Chronic Viral Infections. Front Immunol. 2018;9:276. doi:10.3389/fimmu.2018.00276
63. de Martino M, Lodi L, Galli L, Chiappini E. Immune Response to Mycobacterium tuberculosis: a Narrative Review. Front Pediatr. 2019;7:350. doi:10.3389/fped.2019.00350
64. Jung JH, Rha MS, Sa M, et al. SARS-CoV-2-specific T cell memory is sustained in COVID-19 convalescent patients for 10 months with successful development of stem cell-like memory T cells. Nat Commun. 2021;12(1):4043. doi:10.1038/s41467-021-24377-1
65. Noh JY, Jeong HW, Kim JH, Shin E-C. T cell-oriented strategies for controlling the COVID-19 pandemic. Nat Rev Immunol. 2021;21(11):687–688. doi:10.1038/s41577-021-00625-9
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.