")
Back to Journals » Journal of Blood Medicine » Volume 14
Paroxysmal Nocturnal Hemoglobinuria: Current Management, Unmet Needs, and Recommendations
Authors Oliver M, Patriquin CJ
Received 19 July 2023
Accepted for publication 30 November 2023
Published 6 December 2023 Volume 2023:14 Pages 613—628
DOI https://doi.org/10.2147/JBM.S431493
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Monika Oliver,1 Christopher J Patriquin2
1Department of Medicine, University of Alberta; Division of Hematology, University of Alberta Hospital, Edmonton, Alberta, Canada; 2Department of Medicine (Hematology), University of Toronto, Division of Medical Oncology & Hematology, University Health Network, Toronto, Ontario, Canada
Correspondence: Christopher J Patriquin, Toronto General Hospital, 200 Elizabeth Street, Toronto, Ontario, M5G2C4, Canada, Tel + 1-416-340-5233, Fax + 1-416-340-3799, Email [email protected]
Abstract: Paroxysmal nocturnal hemoglobinuria (PNH) is an ultra-rare, acquired clonal abnormality, which renders hematopoietic cells exquisitely sensitive to complement-mediated destruction. Classical features of PNH include intravascular hemolytic anemia, increased thrombotic risk, and manifestations related to end-organ damage (eg fatigue, chest pain, dyspnea, renal failure, and pulmonary hypertension). With supportive care alone, mortality rate of patients with PNH is approximately 35%. The anti-C5 monoclonal antibody, eculizumab, was the first targeted therapy approved for PNH, and led to improved hemoglobin, quality of life, reduced transfusion need, reduced thrombosis, and greater overall survival. More recently, therapeutics such as longer acting anti-C5 (ravulizumab) and anti-C3 (pegcetacoplan) medications have been approved, along with other novel therapeutics in late-stage clinical trials. Biosimilars of eculizumab are also now available. Proximal inhibitors (against C3, factor B, and factor D) have shown significant improvements in hemoglobin and transfusion-avoidance in patients who remain anemic despite C5 inhibition. Despite these novel therapies, some unmet challenges remain, including management of breakthrough hemolysis, clinically significant iatrogenic extravascular hemolysis, optimal management in pregnancy, and infection risk mitigation as new targets in the complement system are blocked. In addition, the use of self-administered subcutaneous and oral therapies raises concerns around treatment adherence and the risks of uncontrolled terminal complement. Given the ultra-rare nature of PNH, development is underway of a centralized international registry to capture and analyze the data as they mature for various new therapies and characterize the clinical challenges related to PNH management.
Keywords: complement, hemolysis, hemolytic anemia, PNH, thrombosis
Introduction
PNH Pathophysiology and Disease Manifestations
Paroxysmal nocturnal hemoglobinuria (PNH) is an ultra-rare, acquired clonal abnormality of hematopoietic stem cells. The primary defect is a somatic mutation in the PIGA gene leading to the absence of glycosylphosphatidylinositol (GPI) anchors and, therefore, the GPI-anchor-bound molecules in hematopoietic cells. In PNH, the lack of GPI-linked regulatory proteins, CD55 (DAF – decay accelerating factor) and CD59 (MIRL – membrane inhibitor of reactive lysis), drive pathology. In the absence of these regulatory controls, complement activation on the surface of these cells can progress unchecked, culminating in formation of membrane attack complexes. This results in intravascular hemolysis (IVH) of vulnerable erythrocytes and activation of PNH leukocytes and platelets, manifesting in thrombosis risk, systemic morbidity, and pre-mature death.1,2
PNH can affect individuals of any age group but is diagnosed most commonly in the third decade of life. There is no clear geographic, ethnic, or sex predilection. Manifestations of PNH are variable and include fatigue, dyspnea, abdominal pain, hemoglobinuria, and smooth muscle dystonia (eg esophageal spasms and erectile dysfunction). Symptoms are a consequence of unrelenting IVH and the complex interplay of complement and coagulation systems. Life-threatening complications of PNH include chronic kidney disease (CKD), pulmonary hypertension, as well as venous or arterial thromboembolic events (TE).3 Historically, approximately, 20–35% of PNH patient would die within 5–10 years of diagnosis despite best available supportive care.1 Thus, early detection is paramount to allow for prompt initiation of targeted therapy. PNH should be considered in any individual presenting with otherwise unexplained cytopenia, aplastic anemia or other bone marrow failure, atypical thrombotic presentation, Coombs-negative hemolysis, or hemoglobinuria.4 Diagnosis should be via high-sensitivity flow cytometry, to detect GPI-deficient populations of granulocytes, monocytes, and erythrocyte lineages.4,5
Current Standard of Care
Standard of care (SOC) treatment for PNH varies broadly by country, with many nations still without any complement inhibition,6 though this may be improving as clinical trials of novel agents expand geographically. The only curative therapy for PNH is an allogeneic hematopoietic stem cell transplant (HSCT); however, as transplant-related morbidity and mortality are significant, HSCT is reserved for PNH patients with concomitant severe aplastic anemia or other causes of bone marrow failure (BMF), where HSCT is worth the risk of addressing both conditions.1,4,7 However, patients with a history of PNH-related thrombosis appear to do worse with HSCT and are best served by supportive care alone if targeted treatments are not available.8,9 As transplant medicine evolves with less intensive conditioning regimens and improved graft-versus-host disease and infectious prophylaxis, HSCT may eventually move up in the treatment algorithm, especially in jurisdictions where anti-complement therapies are not available. Use of eculizumab prior to transplantation does not appear to affect engraftment or increase transplant complication rates,10 and may protect against increased hemolysis when exposed to antithymocyte globulin.
Currently, the definitive treatment for hemolytic or thrombotic PNH is complement blockade. In Canada, as in many countries, first-line therapy is eculizumab or ravulizumab, monoclonal antibodies targeting terminal complement protein, C5, preventing its cleavage and participation in membrane attack complex formation. With C5 inhibition, IVH is substantially reduced, and major complications are prevented. Details regarding eculizumab and ravulizumab are presented later (see Terminal Complement Inhibition). The C3 inhibitor, pegcetacoplan, has also been given first-line indication in the US,11 though it remains unclear how often it will be chosen before a trial of C5 blockade.
For all patients with PNH, supportive care is essential. This includes hematinic support with folic acid and iron supplementation in iron deficient patients. Transfusion support may be necessary for some patients with severe anemia and, in those patients, iron chelation may be required for transfusion-associated iron overload. Other adjuncts may be used, such as analgesia to manage abdominal pain and smooth muscle dystonia. Prophylactic anticoagulation may be considered for patients at highest thrombotic risk (eg granulocyte clone >30%, ≥2 HDA criteria, and prior TE),12 only if they cannot be started promptly on complement inhibition; however, there are limited data to support this practice and thus a careful assessment of thrombotic and bleeding risk must be undertaken, in discussion with patients.4,13 Both eculizumab and ravulizumab carry a black box warning for life-threatening and fatal meningococcal infections. Thus, all patients require vaccination against meningococcal subtypes. Similar warnings exist for pegcetacoplan.11 Anti-meningococcal antibiotics are required until 14 days post-vaccination, and some patients may elect to continue antimicrobial prophylaxis indefinitely.
Challenges & Unmet Needs
Despite the advances in PNH treatment over the last 20 years, leading to improved clinical outcomes, quality of life (QoL), and overall survival, significant challenges and unmet needs remain. Below, we have framed some of these as they relate to complications of current therapies, convenience of drug administration, and competition in the field of complement-mediated diseases.
Complications
One of the greatest challenges that occurs with current C5 inhibitors is persistent anemia and transfusion dependence. In the phase-3 trial comparing ravulizumab and eculizumab in complement inhibitor-naïve patients (Study 301), 26.4% on ravulizumab and 33.9% on eculizumab remained transfusion-dependent.14 Similarly, in the “switch” trial (Study 302 – ravulizumab versus eculizumab in eculizumab-experienced patients), approximately 24% in each arm did not achieve stabilized hemoglobin (Hb).15 Excluding etiologies unrelated directly to PNH or its treatment,4,9,16 such as bleeding, bone marrow failure (BMF), and hematinic deficiencies, ongoing anemia despite C5 inhibition typically falls into two main categories: patients may have incomplete complement blockade and are susceptible to breakthrough hemolysis (BTH);17,18 or, patients may develop extravascular hemolysis (EVH) of the circulating GPI-negative erythrocytes no longer being hemolyzed intravascularly.19,20
BTH is generally defined as an acute, recurrent elevation in LD plus at least one new or worsening manifestation of intravascular hemolysis (ie hemoglobinuria, fatigue, abdominal pain, TE, dysphagia, erectile dysfunction, or worsening anemia) after a patient has achieved control of IVH (ie LDH <1.5x ULN).21 It may be chronic or recurrent, secondary to insufficient complement inhibition nearing the end of each dosing cycle (pharmacokinetic [PK]-BTH) or regardless of C5 inhibition in the context of complement-amplifying conditions, such as infection or pregnancy (pharmacodynamic [PD]-BTH).18,22–24 A retrospective review of 14 eculizumab-treated patients (763 doses) identified 12 BTH episodes in four patients, most often occurring 24–48 hr before the next scheduled infusion.25 In an analysis of 141 PNH patients treated with eculizumab for ≥13 months, 21% required a higher-than-label dose (ie ≥1200 mg) based on subtherapeutic eculizumab levels or unsuppressed CH50.26 In the phase-3 randomized trials of ravulizumab versus eculizumab, those receiving ravulizumab had a more profound suppression of free C5 and, associated with this, no PK-BTH reported compared to four episodes in the eculizumab arm; PD-BTH occurred in a small number of patients in each group related to infections or other triggers.17,21 Rates and risks of BTH, and how to mitigate them, in patients receiving proximal inhibitors will require careful attention as the data across trials mature (see Proximal Complement Inhibition).
Treatment with C5 inhibition may result in EVH, as the CD55-negative circulating erythrocytes accumulate C3b and its split products. The diagnosis is assumed in the face of a normal or minimally elevated LD accompanied by reticulocytosis and C3d-positive direct antiglobulin test, the latter of which is observed in >50% of PNH patients treated with eculizumab.26,27 A retrospective analysis of 56 PNH patients receiving eculizumab found that 41 (73%) had significant C3 deposition on erythrocytes. The percentage of coated cells correlated with hematologic response, with optimal responders having the lowest C3 deposition.20 These opsonized erythrocytes are tagged for destruction by the reticuloendothelial system and are ultimately hemolyzed within the liver and spleen. Importantly, clinically significant EVH only occurs once a patient is on effective C5 inhibition, and historical treatments including splenectomy or corticosteroids have been ineffective or associated with undue toxicity. In untreated PNH patients, low-level EVH may be theoretically present but is eclipsed by IVH.19 It is not yet possible to accurately identify those patients who may be at higher risk of developing clinically significant EVH, but a contribution of genetic variants in CR1 and complement factor H may play a role.28,29
Even with a suboptimal response to eculizumab or ravulizumab (eg ongoing anemia ± transfusion-dependence), truly uncontrolled disease despite C5 inhibition is rare. Nishimura et al identified a C5 polymorphism in a minority of PNH patients in Japan (3.2%) at a prevalence similar to the Japanese general population (3.5%). This variant leads to a modified binding site such that eculizumab (and ravulizumab) cannot associate. As a result, patients with this C5 polymorphism (p.Arg885His) display frank drug resistance.30
Another complication of therapeutic complement inhibition, which cannot be understated, is infection, particularly due to meningococcus and other encapsulated organisms. Assessing over a decade of post-marketing and pharmacovigilance data on eculizumab use in PNH, the meningococcal infection rate was 0.24 per 100 patient-years, based on a cumulative exposure of 21,016 patient-years.31 Patients require vaccination against meningococcal subtypes (eg ACWY and MenB) at initiation of therapy and every 3–5 years thereafter or based on titers if available; however, immunization alone may not be sufficient, as currently available vaccines do not cover all subtypes. Of the reported cases of meningococcal infection in patients receiving eculizumab in the United States from 2008 to 2016 (14/16 were ACWY-vaccinated), 11/16 N. meningitidis isolates were non-groupable.32 A similar risk exists for gonococcal infection, against which we cannot vaccinate, so careful sexual history and counselling should be done in advance of starting C5 inhibition.33 As novel therapeutics are developed and as different steps in the complement cascade are inhibited, we will need to watch closely for signals of infection risk and work to mitigate them.34
Convenience
Convenience of drug delivery and modality of treatment are also important limitations of currently approved complement inhibitors. Requiring indefinite fortnightly eculizumab infusions is burdensome to patients, caregivers, and healthcare system resources. Ravulizumab improves on this with its longer half-life (49.6 days versus 11.3 days) and is dosed every 8 weeks, allowing more flexibility for patients to travel and have more control of their schedules;14,15 however, intravenous delivery can still be challenging, especially for patients with needle phobias or suboptimal peripheral venous access. A minority may require a peripherally inserted central catheter (PICC) or other implanted device for their infusions.
Novel complement inhibitors have expanded options for delivery, including self-administered subcutaneous (SC) and oral therapies (see Treatment Pathways). SC molecules have variable treatment schedules ranging from multiple times per week,35 to weekly,36 or monthly.37–40 Oral monotherapies currently being tested must be taken twice a day.41,42 Assuming commercial approvals, these options would allow patients to deliver their own treatment, but must be weighed against the risk of poor compliance or inability to tolerate oral intake (eg gastroenteritis), which would expose them to worsening IVH and potential complications, such as TE. The severity of a BTH episode is also potentially increased, with more profound resultant anemia, in patients with larger type-III erythrocyte clones.17 Another consideration is the strict storage requirements for some therapies (eg refrigeration for pegcetacoplan and iptacopan) that may have significant implications for travel. Finally, the potential for drug–drug interactions, not an issue with eculizumab or ravulizumab, would present potential limitations in prescribing and management of comorbid conditions.43
Competition
Widespread access to anti-complement therapy remains a significant challenge. This is driven in large part by the high costs of current SOC therapies, and the economic burden to health-care systems.44–46 Competition in the complement inhibitor space is expected to drive down the benchmark prices of current therapies, while encouraging responsible price listings for new molecules. Prior to commercial listing, multiple industry partners running trials in the same area were driving more rigorous trial methodology, and a more nuanced understanding of the complement system and its inhibition. The latter has the potential to drive the innovation of novel targets to treat not only PNH but also other complement-mediated disorders.47 The frenzy of PNH clinical trials over the past decade has also brought treatment options to countries without prior access to therapies.37,48–50 We remain hopeful that this will result in wider availability and hopefully global reach of effective PNH therapies.
Treatment Pathways
It has now been over 20 years since eculizumab was first used to treat patients with PNH,51 and the field since then has expanded significantly. This is exciting for patients and physicians alike; however, at the same time, many more treatment options can also cause confusion when making clinical decisions. Below, we review the various pathways for therapeutic complement inhibition, focusing on molecules already approved or in late-stage clinical development (see Table 1).
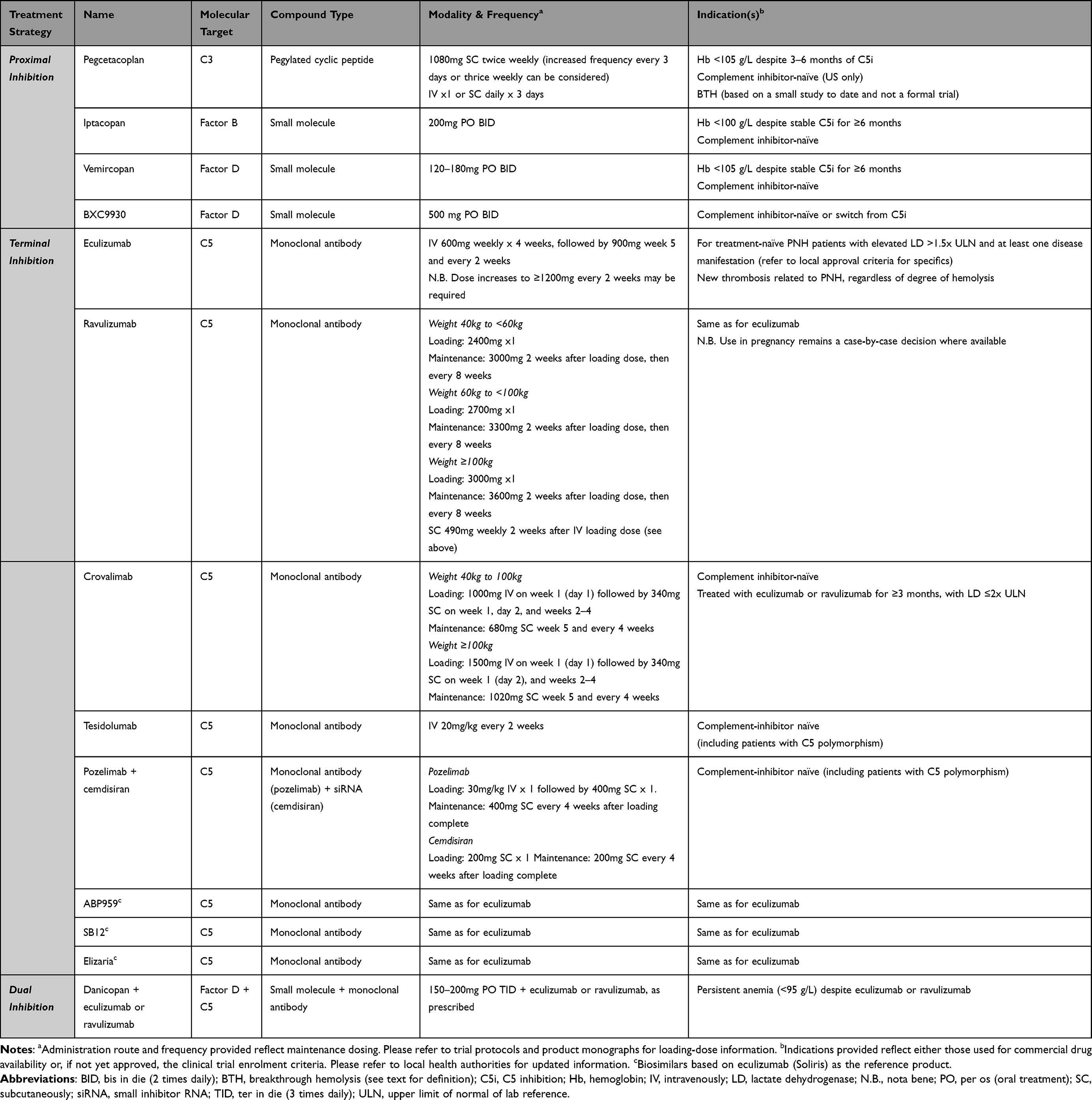 |
Table 1 Complement Inhibitors Approved or in Late-Stage Clinical Trial for Treatment of PNH |
Terminal Complement Inhibition
Eculizumab was the first inhibitor of terminal complement to be developed for clinical use, and even though its benefit in the initially intended indications was underwhelming,47 it has become clear that terminal complement inhibition is highly effective in controlling IVH, and protecting patients from thrombotic and other complications.52 However, approximately 40% of patients receiving eculizumab suffer from significant residual anaemia (Hb <100 g/L) and many remain transfusion dependent.53 It became clear that blocking C5 created the situation of iatrogenic extravascular hemolysis (EVH) due to the persistent circulation of CD55-negative erythrocytes. There are many novel C5 inhibitors, but all are expected to have the same potential for EVH.19
Eculizumab and Ravulizumab
Eculizumab is a fortnightly IV infusion first approved for PNH in 2007. It was the first targeted therapy to show efficacy in the disease, with now over two decades of experience since the first patients enrolled in the pilot study.51 The landmark phase-3 trials (TRIUMPH [N = 87] and SHEPHERD [N = 97]) showed improvement in Hb and transfusion independence, reduction in thrombotic events, recovery of renal function, plus improved fatigue and overall QoL.54–58 Subsequent analysis demonstrated safe and effective use of eculizumab in pregnant patients as well.24
Ravulizumab, like eculizumab, inhibits C5 but offers a half-life approximately four-fold longer, achieved due to a 4-amino-acid substitution; this allows off-loading of C5 in the endosome and recirculation via the neonatal Fc receptor (FcRn).59 Ravulizumab is typically administered by intravenous infusion every 8 weeks, with a weight-based regimen. Efficacy of ravulizumab was shown in two Phase 3 clinical trials (Study 301 [N = 246] and Study 302 [N = 197]).14,15 The primary endpoints of LD improvement or normalization, and transfusion avoidance were achieved. Ravulizumab is noninferior to eculizumab with respect to four key secondary endpoints as well, including the proportion of patients with BTH, Hb stabilization, and improvement in FACIT-fatigue. Subsequent analysis of BTH demonstrated no occurrence of PK-BTH in the ravulizumab arms,21 consistent with the more profound inhibition of free C5.59 Though data need to mature, overall survival for patients receiving ravulizumab also seems similar to those on eculizumab.60 Weekly subcutaneous ravulizumab given via an on-body delivery system has shown similar efficacy, safety, and patient preference,36 and could serve as an alternative to infusions, particularly in situations of poor venous access or during extended travel.
Crovalimab
Crovalimab is a novel C5 inhibitor developed using soluble monoclonal antibody recycling technology (SMART) which, like ravulizumab, offloads bound C5 in acidified endosomes and employs FcRN recycling to extend half-life.61 Following an initial IV loading dose, it is given subcutaneously (SC) every 4 weeks. The COMPOSER trial was a phase-1/2 study of 44 patients who were either naive to treatment or switched from eculizumab at enrolment. It reported continued hemolysis control, Hb stabilization, and transfusion avoidance, with five BTH events reported during the open-label extension phase. Each BTH event occurred in one individual; three of these were temporally associated with an infectious complication, and all resolved with continuation of crovalimab.62 The phase-3 COMMODORE-3 trial, conducted in China, enrolled transfusion-dependent, treatment-naive patients (n=51) with elevated LD (>2× upper limit of normal [ULN]). Both co-primary efficacy endpoints were met, including hemolysis control (LD <1.5× ULN) in 78.7% (95% CI: 67.8–86.6) and transfusion avoidance in 51%.37 Results from the other phase-3 trials, COMMODORE-139 and COMMODORE-2,38 have recently been reported for patients who were either switched from SOC or were treatment-naive. Similar control of hemolysis and transfusion needs were seen. Of note, in the switch trial, 16% of crovalimab-treated patients had drug-target-drug complex (DTDC)-mediated Type III hypersensitivity events occur due to the two anti-C5 antibodies targeting separate binding sites. Most events were mild-to-moderate, isolated to skin and joints, and resolved with no change in crovalimab dosing.39
Pozelimab + Cemdisiran
A phase-3, randomized trial of the anti-C5 monoclonal, pozelimab,63 plus cemdisiran,64 compared to SOC (ravulizumab or eculizumab) is currently recruiting at the time of writing. In addition to directly inhibiting C5 with pozelimab, the use of cemdisiran, a small interfering RNA (siRNA), should further reduce terminal complement activity by directly suppressing hepatic C5 synthesis. Cemdisiran alone has shown a >90% reduction of C5 production in patients with PNH; however, this was insufficient to fully inhibit complement activation alone. The two-drug combination, as part of a phase-2 trial, demonstrated successful control of terminal complement, with an interim analysis (n=16) demonstrating that most participants maintained normalization of LD, and 75% achieved Hb stabilization.40 Results of the phase-3 trial are anticipated.
Tesidolumab
Tesidolumab is a fully human anti-C5 monoclonal antibody, currently used mostly in Asia. Like crovalimab and pozelimab, it too binds to a distinct epitope from eculizumab and ravulizumab, permitting use in those with C5 polymorphisms.30 A phase-2 trial of nine patients, five of whom carrying the Arg885His C5 variant, demonstrated approximately 80% reduction in LD levels, decreased transfusion dependence, a 20 g/L mean Hb increase, and improvement in FACIT-fatigue scores.65 Further, publicly available data are awaited.
Eculizumab Biosimilars
There are currently three eculizumab biosimilars,66 and their reduced cost should allow for expanded availability of complement inhibition in jurisdictions currently still without these life-saving therapies. Two of the molecules, ABP-95967,68 and SB1249,69 are either recently available in some countries or are in the process of being assessed by national health authorities. They have shown comparable efficacy, safety, and immunogenicity to eculizumab as a reference product. Elizaria is another eculizumab biosimilar, currently marketed in Russia, and is a first-line therapy for patients with PNH. Those previously on eculizumab were subsequently switched once it was available.70 A phase-3 trial confirmed non-inferiority compared to eculizumab with respect to LD control, secondary outcomes, and safety parameters.48 How these biosimilars, given fortnightly, will fit into health systems that already have ravulizumab remains to be seen, but could at least provide a more affordable option for the many countries still without C5 inhibition for PNH. To our knowledge, there are no biosimilars that are currently longer acting.
Proximal Complement Inhibition
Interest in proximal complement inhibition as a therapeutic strategy stems from the need to correct the iatrogenic C3-mediated EVH that can occur in PNH patients on a C5 inhibitor. Much of the early work came from the Lambris lab with the C3 inhibitor, compstatin.71 Its pegylated version, pegcetacoplan, was the first such therapeutic approved for use in patients with PNH. Three main classes of proximal inhibitor exist and are either approved for use or are in late-stage clinical trials, blocking C3, factor D (FD), and factor B (FB).72
C3 Inhibition
C3 inhibition is the first successful strategy to treat PNH since terminal complement inhibitors were approved. Pegcetacoplan, a pegylated anti-C3 cyclic peptide, given as a twice-weekly SC infusion, demonstrated superiority in change from baseline Hb (mean difference 38.4 g/L) compared to eculizumab in a randomized, open-label clinical trial (PEGASUS) of patients who had persistent anemia (Hb <105 g/L) despite eculizumab, many of whom were transfusion-dependent.35 It was found to be non-inferior in terms of transfusion-avoidance and change from baseline reticulocyte count, but not for change from baseline LD. Due to hierarchical statistical analysis, QoL was not formally analyzed; however, a post hoc analysis found pegcetacoplan superior to eculizumab across FACIT-fatigue domains.73
Following PEGASUS, PRINCE was an open-label, controlled trial completed in complement inhibitor-naive patients, compared to supportive care alone (ie no C5 inhibition).50 Pegcetacoplan was superior with respect to Hb stabilization (85.7% vs 0% in the SOC arm) and hemolysis control, with a mean LD at 26 weeks of 205 U/L vs 1535 U/L (ULN = 226 U/L). Patients in any of the parent pegcetacoplan trials were eligible to be enrolled in the APL2-307 open-label extension study of pegcetacoplan and demonstrated durable improvements in Hb, transfusion avoidance, LD, and FACIT-fatigue scores.74
A key finding of the pegcetacoplan trials was a significant expansion of GPI-negative erythrocyte populations, often exceeding 90%. This was associated with the increased Hb seen; however, in PEGASUS, episodes of acute hemolysis occurred in four patients and were dramatic, with LD increasing to 10–15x ULN in some cases, and 3 out of the 4 participants discontinued pegcetacoplan.35 Recognition of the potential for profound BTH prompted a sub-study of those in the open extension period, in which those with acute hemolysis could receive either a single IV dose, or three consecutive-day doses of SC pegcetacoplan, followed by every-3-days dosing. Preliminary data suggest that this strategy can successfully manage BTH in many patients on pegcetacoplan,75 though some may benefit from temporary use of C5 inhibitors.76 In the APL2-307 extension trial, 16.8% of patients developed an acute hemolytic episode. All of these resolved, though 7/9 received increased dosing and three required transfusions. Three of these patients elected to discontinue trial participation due to hemolysis.74 A clear and stratified approach on how best to manage BTH for physicians will be essential.
Factor B Inhibition
Factor B is an enzyme only active in the alternative pathway of complement and is required to form the alternative-pathway convertases of C3 and C5. Targeting FB theoretically permits ongoing functionality of the classical and lectin pathways. Iptacopan is an orally available, small-molecule FB inhibitor being trialed in multiple complement-mediated conditions. Its activity in PNH was first demonstrated as an add-on therapy for those still anemic despite C5 inhibition.77 In the phase-3 APPLY-PNH trial of anemic patients (n=97) despite eculizumab, twice daily iptacopan monotherapy was associated with a mean Hb increase of 35.9 g/L. Only two patients in the iptacopan arm received transfusions versus 60% of those who stayed on eculizumab.42 The APPOINT-PNH trial is a single-arm study of complement inhibitor-naive patients (n=40) with baseline Hb <100 g/L. In a press release, Novartis announced that, at 24 weeks, 92.2% of patients had a ≥20 g/L increase in Hb, 62.8% had a Hb of 120 g/L or more, and 95% had LD below 1.5x ULN.78,79
Factor D Inhibition
Factor D is synthesized primarily in adipose tissue and circulates at the lowest concentration of all complement proteins.80 This is the rate-limiting step of the alternative pathway, as it needs to cleave FB for it to partake in convertase activity with C3b or C3H2O. As with FB blockade, inhibition of FD impairs alternative pathway complement function while allowing other pathways to function. Danicopan, discussed in more detail later (see Dual-Target Inhibition), was the first FD inhibitor to show activity in PNH patients and provided a proof of concept. Since then, vemircopan and BCX9930 have moved forward in trials, used as oral monotherapy.
Vemircopan, taken twice daily, is advancing through a phase-2 clinical trial program with three cohorts, including those who have switched from danicopan monotherapy, those with suboptimal response to C5 inhibition, and treatment-naive patients. In the last group, 54.5% of whom were receiving transfusions during screening, vemircopan was associated with a mean Hb increase of 39 g/L, 81% reduction in LD, clinically significant improvement in FACIT-fatigue scores, and only one patient required transfusion on day 2 of treatment.41 Updated data and results from the other cohorts are anticipated.
BCX9930 is another twice-daily oral FD inhibitor that was being tested in patients with PNH who were either suboptimal responders to C5 inhibition or were treatment-naive. Those receiving the drug showed improved Hb and hemolytic parameters and reduced transfusion requirements.81,82 Due to concerns related to crystalline nephropathy, potentially limiting dose optimization, the program was discontinued; however, patients already on BCX9930 and benefiting from it have continued. In a press release, BioCryst announced that they are in the early stages of developing a once-daily FD inhibitor (BCX10013) for another attempt at treating PNH and other complement-mediated diseases.83
Dual-Target Inhibition
The current rationale for dual-target complement inhibition in PNH is to maintain a direct blockade of C5 while also providing proximal inhibition to protect against or treat EVH. Proximal inhibition would in essence allow for greater increments in Hb, while terminal blockade could protect against severe episodes of BTH in patients with significant proportions of circulating type-III erythrocytes.17
In eculizumab-treated PNH patients who remained transfusion-dependent, presumably due to EVH, the addition of thrice-daily danicopan (anti-FD) in a phase-2 trial was associated with a mean Hb increase of 24 g/L and a significant reduction in transfusion needs.84 The phase-3 ALPHA trial of the same combination has completed enrolment, with full results anticipated soon.43 Rapid increases in Hb were similarly seen in the PEGASUS trial during the 4-week run-in period, where participants received both pegcetacoplan and eculizumab.35 A trial of KP104, a bifunctional monoclonal antibody that inhibits C5 while employing complement factor H activity for proximal pathway regulation, has recently started in patients with PNH, with data eagerly awaited.85
A combined-target approach to treating PNH is certainly intriguing, and the data thus far suggest that it could be successful. The main limiting factor will be economic. This could be possible if both therapeutics are from the same company or, in the example of KP104, a single agent has multiple targets; however, given the extreme prices of orphan drugs, even modest incremental cost may be infeasible. Beyond pricing, questions remain surrounding who would benefit most, if combination therapy is required long term or for certain periods, and which combinations are most safe and effective.
Management Recommendations
The goal of the International PNH Interest Group (IPIG - https://www.pnhinterestgroup.org/) and other organizations is to continue to advocate for effective therapies for PNH patients internationally. Considering this important goal, we have outlined some clinical scenarios and topics that may be of help to physicians who may not manage PNH on a regular basis. We would also encourage the reader to refer to national standards and guideline documents where available.
Initiating Therapy
In patients with small clones (usually granulocyte populations <10%) with no evidence of hemolysis or PNH-related disease manifestations, anti-complement therapy may not be required and it may be appropriate to closely monitor with blood work and clinical assessments. Close monitoring and supportive care alone may also be sufficient in patients with larger clones who do not otherwise meet indications for targeted therapy.
In patients with clinically significant hemolytic PNH, control of terminal complement is central, irrespective of the therapeutic target chosen. This is also the case for thrombotic PNH which can in some cases present without significant hemolysis, particularly if there is a predominance of type-II erythrocytes.86 Though this may change soon, in Canada we remain limited to eculizumab and, in some situations, ravulizumab. Many jurisdictions use high disease activity (HDA)87 criteria for approval decisions, permitting any PNH patient with LD >1.5x ULN and at least one manifestation of the disease to receive therapy. Some countries, such as Canada, may be limited by more restrictive criteria that are set by national HTA bodies.4,88 In addition to standard approval criteria, pregnancy in a patient with PNH is another indication to start eculizumab, even if they were not receiving C5 inhibition prior to conception, given the increased risk during this time.24 In some urgent situations, such as new TE or severe hemolysis with end-organ damage (eg acute kidney injury), C5 inhibition must be started urgently to prevent progression. The rapidity of terminal complement blockade is crucial in these scenarios. As such, we would not use subcutaneous therapies, which can take longer to reach therapeutic levels.89 Particularly with these presentations, we would not delay starting drug due to vaccination, and would cover patients with anti-meningococcal prophylaxis until it is possible and safe to vaccinate (see Mitigating Infection Risk).4 In the case of acute thrombosis, therapeutic anticoagulation is strongly recommended. There is little data to guide choice or duration of anticoagulation for TE in the setting of PNH, and it would depend in part on the site and severity of the TE presentation; however, we will routinely use direct oral anticoagulants (DOACs) for a minimum of 3–6 months and until control of IVH is achieved after starting anti-complement therapy. We engage in shared decision-making with patients thereafter, to weigh recurrent thrombotic versus bleeding risk factors to determine the need for ongoing prophylaxis thereafter.
At the time of writing, based on the evidence available, we would suggest starting patients on C5 inhibition in the upfront setting, with ravulizumab being preferred due to convenience and lower risk of PK BTH.21 This may change over time, as naïve-start data for other therapeutics mature; the PRINCE and APPOINT-PNH data suggest that pegcetacoplan and iptacopan, respectively, are also safe and effective therapies to start in complement inhibitor-naive individuals.50,79 With access to multiple anti-complement therapies, patient preference will be a key consideration. In jurisdictions with limited access, we would favour initiating complement blockade in any form available given the risks of untreated PNH.1,52
Whom to Switch
Patients who have ongoing anemia attributed to clinically significant EVH, particularly if symptomatic or transfusion-dependent, would be the optimal candidates to be switched to proximal inhibition. The choice of therapeutics will depend on patient preference, clinical characteristics, and jurisdictional availability. Most therapies recently approved or in late-stage development have clinical trial data for patients switching from SOC C5 inhibition. At baseline, patients who meet the characteristics of trial participants are those whom we would consider switching (eg persistent anemia despite 3–6 months of C5 inhibition).
Criteria to gauge hematologic response have been proposed, which consider Hb, LD, and reticulocyte count.19 A subsequent multicentre review (n=160) found that 21.3% had a complete response (normal Hb, transfusion-independent), 40.2% had a good response (Hb 100–120 g/L, transfusion-independent), with the remainder achieving only partial (26.8%) or minor (11.8%) response.53 However, these criteria have been criticized for valuing an increased Hb over LD, a marker of IVH (eg a “major response” would permit LD >1.5x ULN, as long as the Hb is over 120 g/L).90 Being permissive of LD >1.5x ULN could place patients at risk of TE, as previous studies have shown a direct association between elevated LD and thrombosis.91 Another concern is that regulators and payers could withdraw life-saving therapies if improvement in Hb is weighted too heavily, without consideration of LD control. As such, a re-evaluation of response criteria is planned.
With respect to switching treatments, there are further considerations, not least of which is patient preference and willingness to change. As deep and consistent complement blockade is necessary to protect patients from TE and end-organ damage, additional issues such as adherence to oral therapies41,42,92 or challenges with self-administered delivery mechanisms (eg syringes and infusion pumps)35–37,62,93 need to be carefully considered.
Based on the data thus far, those with a significant thrombotic history seem best served by staying on a C5 monoclonal antibody; in particular, there are no data that we are aware of to support starting a novel therapeutic in a patient presenting with de novo TE. Lastly, we must be very cautious in the context of pregnancy since pregnant and nursing patients were explicitly excluded from trials. For now, we would recommend remaining on (or switching to) eculizumab during pregnancy given the long-term experience and data demonstrating its beneficial use,24 though there are different practices and opinions, and this may change as safety data become available.
Mitigating Infection Risk
All patients receiving complement inhibition to treat PNH require meningococcal vaccination, with both quadrivalent and serogroup B formulations where available. As vaccines can amplify complement activity and lead to acute hemolysis,94 it is our standard practice to first start complement inhibition and administer vaccines thereafter, providing antimicrobial prophylaxis for at least 14 days after they are given. For long-term protection, we provide boosters for each vaccine every 3 years (maximum, every 5 years).4 Others, with the assay available, will revaccinate at a variable frequency based on anti-meningococcal titres. Pneumococcal and Haemophilus type B vaccinations are recommended for patients receiving proximal inhibitors. Despite concerns that blocking “higher up” the complement cascade could increase infection risk, this has not been seen to date in the data available. Indeed, response to meningococcal vaccination was no worse in those patients receiving danicopan compared to those on eculizumab.95,96 As data mature for other therapies, we will need to watch vigilantly for any safety signals of increased or differential infection risk.
Long-term prophylactic antibiotics in addition to vaccination are recommended in several countries, but not all. The most common regimen is penicillin twice-daily, or possibly azithromycin in the case of penicillin allergy.4 An analysis of the International PNH Registry did not find any significant difference in rate of meningococcal infection between those who did and did not take prophylactic antibiotics (0.1 per 100 patient-years), but the analysis was limited by lack of a standard definition and duration of “prophylactic” and low meningococcal infection event rates.97 The decision to take long-term antibiotics depends on patient values and preferences, and the possibility of side effects. Irrespective of prophylactic antimicrobial use, many experts suggest a “pill in pocket” approach, in which patients fill a prescription (typically of ciprofloxacin) with instructions to take a dose at the first signs of meningitis or unexplained fever and proceed to their closest emergency department. This is particularly important for patients who may live in remote areas or when abroad. Use of eculizumab without immunization has been done and reported safe, but only in the context of HSCT-associated thrombotic microangiopathy, during the discrete period before immune reconstitution.98 It is rare to have a patient with PNH unable to receive vaccines; however, a discussion of the risks and benefits of complement inhibition without immunization would need to occur, and antimicrobial prophylaxis would be necessary.
Pregnancy
Management of PNH patients who become pregnant remains an important focus. Without complement inhibition, maternal mortality, mostly driven by thrombotic complications, ranged from 8% to 20% historically.24 There is now many years’ experience using eculizumab in this context, with improved maternal outcomes and acceptable maternal and neonatal safety profile.24 For our pregnant patients, we also recommend low molecular weight heparin prophylaxis during pregnancy and for 6 weeks post-partum, as well as early initiation of aspirin to protect against preeclampsia. All of these patients are managed alongside colleagues in high-risk obstetrics. There is growing experience using ravulizumab in pregnancy, but few published reports; the implications of greater reliance on the FcRn on the fetus require further attention. The safety of other complement inhibitors remains to be seen, and there is a plan in place to do this centrally within the IPIG Registry.
Where are We Going?
The PNH treatment landscape is evolving. With new treatments on the horizon, patients and physicians alike are excited about the trajectory and momentum of this evolution. Unfortunately, targeted anti-complement agents are still available in only a minority of countries.6 With increased competition in terms of PNH treatments, we are hopeful that the cost of SOC therapy will be reduced; however, as with most orphan drugs, we anticipate that costs will continue to be high.45,46,99 This will create challenges for resource-limited countries and may result in delays in drug approval.
As new therapies become available, and hopefully have broader geographic spread, it will be important to capture efficacy and safety data comprehensively. The current International PNH Registry (NCT01374360) was established in 2007, when eculizumab was the only treatment available and has been an invaluable resource to advance our knowledge of PNH and its management. A new registry, under the auspices of the International PNH Interest Group, is planned to start in the near future and will aim to enroll PNH patients receiving therapeutics by any company that participates, as well as those patients not on complement inhibition.
As the field progresses, and long-term extension data for newer therapies mature, prescribers will require a roadmap to help navigate the anti-complement space, including direction on initiation in treatment-naive patients, whom and when to switch, and how to manage specific situations like BTH, TE, and pregnancy. International guidelines that will set standards for overall diagnostic and management strategies, while accounting for jurisdictional variation, will be an invaluable resource. Guidance should also come from local consensus guidelines, which consider the available therapies and nuances in each region. Physicians and patients will need to engage in shared decision-making to determine which therapy fits their disease characteristics and lifestyle needs. Drug availability, compliance, and modality of delivery will be important pillars of these conversations.
While the gaps in the PNH treatment landscape are reducing, there remain several unmet needs as we have outlined here. A more nuanced and precision medicine-based understanding of PNH could identify patients at particular risk of incomplete IVH control or clinically significant EVH with C5 inhibition. This information may allow us to risk-stratify patients at the time of diagnosis, and guide whether distal or proximal complement inhibition is most appropriate in the front-line setting, and which proximal target would be most effective. Lastly, additional complement biomarkers would be welcome, allowing a clearer distinction between IVH and EVH, and more objective targets to guide when a switch in therapy should be considered.100 We have learnt much over the last 20 years since targeted complement inhibition first became possible, with great strides toward improvements in patient care. Given the ultra-rare nature of PNH, ongoing success now and in the future will come from continued and expanded international collaboration.
Author Contributions
Both authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
Dr Monika Oliver reports personal fees from Alexion, Sobi, Novartis, Takeda, and Sanofi, outside the submitted work. Dr Christopher Patriquin reports personal fees from Alexion, BioCryst, Amgen, Roche, Novartis, Sobi, Sanofi, and Takeda; Global PI for PNH clinical trial for Regeneron, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1995;333(19):1253–1258. doi:10.1056/NEJM199511093331904
2. Brodsky RA. Narrative review: paroxysmal nocturnal hemoglobinuria: the physiology of complement-related hemolytic anemia. Ann Intern Med. 2008;148(8):587–595. doi:10.7326/0003-4819-148-8-200804150-00003
3. Schrezenmeier H, Muus P, Socie G, et al. Baseline characteristics and disease burden in patients in the international paroxysmal nocturnal hemoglobinuria registry. Haematologica. 2014;99(5):922–929. doi:10.3324/haematol.2013.093161
4. Patriquin CJ, Kiss T, Caplan S, et al. How we treat paroxysmal nocturnal hemoglobinuria: a consensus statement of the Canadian PNH Network and review of the national registry. Eur J Haematol. 2019;102(1):36–52. doi:10.1111/ejh.13176
5. Borowitz MJ, Craig FE, Digiuseppe JA, et al. Guidelines for the diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria and related disorders by flow cytometry. Cytometry B Clin Cytom. 2010;78(4):211–230. doi:10.1002/cyto.b.20525
6. Partnering4PNH. A global policy consensus paper to address remaining unmet medical needs for people living with PNH; 2022. Available from: https://pnh.sobi.com/sites/default/files/P4PNH-consensus-paper-short-version%20-%2015.12.2022%20.pdf.
7. Brodsky RA. Stem cell transplantation for paroxysmal nocturnal hemoglobinuria. Haematologica. 2010;95(6):855–856. doi:10.3324/haematol.2010.023176
8. Peffault de Latour R, Schrezenmeier H, Bacigalupo A, et al. Allogeneic stem cell transplantation in paroxysmal nocturnal hemoglobinuria. Haematologica. 2012;97(11):1666–1673. doi:10.3324/haematol.2012.062828
9. Risitano AM, Peffault de Latour R. How we(‘ll) treat paroxysmal nocturnal haemoglobinuria: diving into the future. Br J Haematol. 2022;196(2):288–303. doi:10.1111/bjh.17753
10. Vallet N, de Fontbrune FS, Loschi M, et al. Hematopoietic stem cell transplantation for patients with paroxysmal nocturnal hemoglobinuria previously treated with eculizumab: a retrospective study of 21 patients from SFGM-TC centers. Haematologica. 2018;103(3):e103–e105. doi:10.3324/haematol.2017.182360
11. Heo YA. Pegcetacoplan: a review in paroxysmal nocturnal haemoglobinuria. Drugs. 2022;82(18):1727–1735. doi:10.1007/s40265-022-01809-w
12. Hochsmann B, Peffault de Latour R, Hill A, et al. Risk factors for thromboembolic events in patients with paroxysmal nocturnal hemoglobinuria (PNH): a nested case-control study in the International PNH Registry. Ann Hematol. 2023;102(11):2979–2988. doi:10.1007/s00277-023-05402-3
13. Griffin M, Munir T. Management of thrombosis in paroxysmal nocturnal hemoglobinuria: a clinician’s guide. Ther Adv Hematol. 2017;8(3):119–126. doi:10.1177/2040620716681748
14. Lee JW, Sicre de Fontbrune F, Wong Lee Lee L, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood. 2019;133(6):530–539. doi:10.1182/blood-2018-09-876136
15. Kulasekararaj AG, Hill A, Rottinghaus ST, et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor-experienced adult patients with PNH: the 302 study. Blood. 2019;133(6):540–549. doi:10.1182/blood-2018-09-876805
16. Kulasekararaj AG, Brodsky RA, Hill A. Monitoring of patients with paroxysmal nocturnal hemoglobinuria on a complement inhibitor. Am J Hematol. 2021;96(7):E232–E235. doi:10.1002/ajh.26176
17. Notaro R, Luzzatto L. Breakthrough Hemolysis in PNH with Proximal or Terminal Complement Inhibition. N Engl J Med. 2022;387(2):160–166. doi:10.1056/NEJMra2201664
18. Brodsky RA. How I treat paroxysmal nocturnal hemoglobinuria. Blood. 2021;137(10):1304–1309. doi:10.1182/blood.2019003812
19. Risitano AM, Marotta S, Ricci P, et al. Anti-complement treatment for paroxysmal nocturnal hemoglobinuria: time for proximal complement inhibition? A position paper from the SAAWP of the EBMT. Front Immunol. 2019;10:1157. doi:10.3389/fimmu.2019.01157
20. Risitano AM, Notaro R, Marando L, et al. Complement fraction 3 binding on erythrocytes as additional mechanism of disease in paroxysmal nocturnal hemoglobinuria patients treated by eculizumab. Blood. 2009;113(17):4094–4100. doi:10.1182/blood-2008-11-189944
21. Brodsky RA, Peffault de Latour R, Rottinghaus ST, et al. Characterization of breakthrough hemolysis events observed in the phase 3 randomized studies of ravulizumab versus eculizumab in adults with paroxysmal nocturnal hemoglobinuria. Haematologica. 2021;106(1):230–237. doi:10.3324/haematol.2019.236877
22. Harder MJ, Kuhn N, Schrezenmeier H, et al. Incomplete inhibition by eculizumab: mechanistic evidence for residual C5 activity during strong complement activation. Blood. 2017;129(8):970–980. doi:10.1182/blood-2016-08-732800
23. Miyasaka N, Miura O, Kawaguchi T, et al. Pregnancy outcomes of patients with paroxysmal nocturnal hemoglobinuria treated with eculizumab: a Japanese experience and updated review. Int J Hematol. 2016;103(6):703–712. doi:10.1007/s12185-016-1946-x
24. Kelly RJ, Hochsmann B, Szer J, et al. Eculizumab in pregnant patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2015;373(11):1032–1039. doi:10.1056/NEJMoa1502950
25. Nakayama H, Usuki K, Echizen H, Ogawa R, Orii T. Eculizumab dosing intervals longer than 17 days may be associated with greater risk of breakthrough hemolysis in patients with paroxysmal nocturnal hemoglobinuria. Biol Pharm Bull. 2016;39(2):285–288. doi:10.1248/bpb.b15-00703
26. McKinley CE, Richards SJ, Munir T, et al. Extravascular hemolysis due to C3-loading in patients with PNH treated with eculizumab: defining the clinical syndrome. Blood. 2017;130(1):3471.
27. Hill A, Rother RP, Arnold L, et al. Eculizumab prevents intravascular hemolysis in patients with paroxysmal nocturnal hemoglobinuria and unmasks low-level extravascular hemolysis occurring through C3 opsonization. Haematologica. 2010;95(4):567–573. doi:10.3324/haematol.2009.007229
28. Rondelli T, Risitano AM, Peffault de Latour R, et al. Polymorphism of the complement receptor 1 gene correlates with the hematologic response to eculizumab in patients with paroxysmal nocturnal hemoglobinuria. Haematologica. 2014;99(2):262–266. doi:10.3324/haematol.2013.090001
29. Prata PH, Galimard JE, Sicre de Fontbrune F, et al. Rare germline complement factor H variants in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2023;141(15):1812–1816. doi:10.1182/blood.2022017019
30. Nishimura J, Yamamoto M, Hayashi S, et al. Genetic variants in C5 and poor response to eculizumab. N Engl J Med. 2014;370(7):632–639. doi:10.1056/NEJMoa1311084
31. Socie G, Caby-Tosi MP, Marantz JL, et al. Eculizumab in paroxysmal nocturnal haemoglobinuria and atypical haemolytic uraemic syndrome: 10-year pharmacovigilance analysis. Br J Haematol. 2019;185(2):297–310. doi:10.1111/bjh.15790
32. McNamara LA, Topaz N, Wang X, Hariri S, Fox L, MacNeil JR. High risk for invasive meningococcal disease among patients receiving Eculizumab (Soliris) despite receipt of meningococcal vaccine. MMWR Morb Mortal Wkly Rep. 2017;66(27):734–737. doi:10.15585/mmwr.mm6627e1
33. Crew PE, Abara WE, McCulley L, et al. Disseminated gonococcal infections in patients receiving eculizumab: a case series. Clin Infect Dis. 2019;69(4):596–600. doi:10.1093/cid/ciy958
34. Heesterbeek DAC, Angelier ML, Harrison RA, Rooijakkers SHM. Complement and bacterial infections: from molecular mechanisms to therapeutic applications. J Innate Immun. 2018;10(5–6):455–464. doi:10.1159/000491439
35. Hillmen P, Szer J, Weitz I, et al. Pegcetacoplan versus Eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2021;384(11):1028–1037. doi:10.1056/NEJMoa2029073
36. Yenerel MN, Sicre de Fontbrune F, Piatek C, et al. Phase 3 study of subcutaneous versus intravenous ravulizumab in eculizumab-experienced adult patients with PNH: primary analysis and 1-year follow-up. Adv Ther. 2023;40(1):211–232. doi:10.1007/s12325-022-02339-3
37. Liu H, Xia L, Weng J, et al. Efficacy and safety of the C5 inhibitor crovalimab in complement inhibitor-naive patients with PNH (COMMODORE 3): a multicenter, Phase 3, single-arm study. Am J Hematol. 2023;98(9):1407–1414. doi:10.1002/ajh.26998
38. Röth A, He G, Brodsky A, et al. The Phase III, randomized commodore 2 trial: results from a multicenter study of crovalimab vs eculizumab in paroxysmal nocturnal hemoglobinuria (pnh) patients naïve to complement inhibitors.
39. Scheinberg P, Cle D, Edwards J, et al. PHASE III randomized, multicenter, open-label commodore 1 trial: comparison of crovalimab vs eculizumab in complement inhibitor-experienced patients with Paroxysmal Nocturnal Hemoglobinuria (PNH).
40. Devalaraja-Narashimha K, Huang C, Cao M, et al. Pharmacokinetics and pharmacodynamics of pozelimab alone or in combination with cemdisiran in non-human primates. PLoS One. 2022;17(6):e0269749. doi:10.1371/journal.pone.0269749
41. Browett PJ, Kulasekararaj A, Notaro R, et al. Vemircopan (ALXN2050) monotherapy in paroxysmal nocturnal hemoglobinuria: interim data from a Phase 2 open-label proof-of-concept study. Blood. 2022;140(2):717–719. doi:10.1182/blood-2022-169301
42. de Latour RPRA, Kulasekararaj A, Scheinberg P, et al. Oral monotherapy with iptacopan, a proximal complement inhibitor of factor B, has superior efficacy to intravenous terminal complement inhibition with standard of care eculizumab or ravulizumab and favorable safety in patients with paroxysmal nocturnal hemoglobinuria and residual anemia: results from the randomized, active-comparator-controlled, open-label, multicenter, phase III apply-PNH Study. Blood. 2022;140(2). doi:10.1182/blood-2022-171469
43. Kulasekararaj AG, Lazana I. Paroxysmal nocturnal hemoglobinuria: where are we going. Am J Hematol. 2023;98 Suppl 4:S33–S43. doi:10.1002/ajh.26882.
44. Bektas M, Copley-Merriman C, Khan S, Sarda SP, Shammo JM. Paroxysmal nocturnal hemoglobinuria: patient journey and burden of disease. J Manag Care Spec Pharm. 2020;26(12–b Suppl):S8–S14. doi:10.18553/jmcp.2020.26.12-b.s8
45. Rizzardo S, Bansback N, Dragojlovic N, et al. Evaluating Canadians’ Values for Drug Coverage Decision Making. Value Health. 2019;22(3):362–369. doi:10.1016/j.jval.2018.08.008
46. Lancet Haematology T. The rising cost of orphan drugs. Lancet Haematol. 2015;2(11):e456. doi:10.1016/S2352-3026(15)00229-X
47. Patriquin CJ, Kuo KHM. Eculizumab and beyond: the past, present, and future of complement therapeutics. Transfus Med Rev. 2019;33(4):256–265. doi:10.1016/j.tmrv.2019.09.004
48. Kulagin AD, Ptushkin VV, Lukina EA, et al. Randomized multicenter noninferiority phase III clinical trial of the first biosimilar of eculizumab. Ann Hematol. 2021;100(11):2689–2698. doi:10.1007/s00277-021-04624-7
49. Jang JH, Gomez RD, Bumbea H, et al. A phase III, randomised, double-blind, multi-national clinical trial comparing SB12 (proposed eculizumab biosimilar) and reference eculizumab in patients with paroxysmal nocturnal haemoglobinuria. EJHaem. 2023;4(1):26–36. doi:10.1002/jha2.632
50. Wong RSM, Navarro-Cabrera JR, Comia NS, et al. Pegcetacoplan controls hemolysis in complement inhibitor-naive patients with paroxysmal nocturnal hemoglobinuria. Blood Adv. 2023;7(11):2468–2478. doi:10.1182/bloodadvances.2022009129
51. Hillmen P, Hall C, Marsh JC, et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2004;350(6):552–559. doi:10.1056/NEJMoa031688
52. Kulasekararaj AG, Brodsky RA, Nishimura JI, Patriquin CJ, Schrezenmeier H. The importance of terminal complement inhibition in paroxysmal nocturnal hemoglobinuria. Ther Adv Hematol. 2022;13:20406207221091046. doi:10.1177/20406207221091046
53. Debureaux PE, Kulasekararaj AG, Cacace F, et al. Categorizing hematological response to eculizumab in paroxysmal nocturnal hemoglobinuria: a multicenter real-life study. Bone Marrow Transplant. 2021;56(10):2600–2602. doi:10.1038/s41409-021-01372-0
54. Kelly RJ, Hill A, Arnold LM, et al. Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood. 2011;117(25):6786–6792. doi:10.1182/blood-2011-02-333997
55. Loschi M, Porcher R, Barraco F, et al. Impact of eculizumab treatment on paroxysmal nocturnal hemoglobinuria: a treatment versus no-treatment study. Am J Hematol. 2016;91(4):366–370. doi:10.1002/ajh.24278
56. Hillmen P, Muus P, Duhrsen U, et al. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007;110(12):4123–4128. doi:10.1182/blood-2007-06-095646
57. Hillmen P, Young NS, Schubert J, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233–1243. doi:10.1056/NEJMoa061648
58. Brodsky RA, Young NS, Antonioli E, et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;111(4):1840–1847. doi:10.1182/blood-2007-06-094136
59. Peffault de Latour R, Brodsky RA, Ortiz S, et al. Pharmacokinetic and pharmacodynamic effects of ravulizumab and eculizumab on complement component 5 in adults with paroxysmal nocturnal haemoglobinuria: results of two phase 3 randomised, multicentre studies. Br J Haematol. 2020;191(3):476–485. doi:10.1111/bjh.16711
60. Kulasekararaj A, Brodsky R, Griffin M, et al. Long-term complement inhibition and survival outcomes in patients with paroxysmal nocturnal hemoglobinuria: an interim analysis of the ravulizumab clinical trials. Hemasphere. 2022;6:706–707. doi:10.1097/01.HS9.0000846132.33920.32
61. Roth A, Nishimura JI, Nagy Z, et al. The complement C5 inhibitor crovalimab in paroxysmal nocturnal hemoglobinuria. Blood. 2020;135(12):912–920. doi:10.1182/blood.2019003399
62. Roth A, Ichikawa S, Ito Y, et al. Crovalimab treatment in patients with paroxysmal nocturnal haemoglobinuria: long-term results from the phase I/II COMPOSER trial. Eur J Haematol. 2023;111(2):300–310. doi:10.1111/ejh.14011
63. Latuszek A, Liu Y, Olsen O, et al. Inhibition of complement pathway activation with Pozelimab, a fully human antibody to complement component C5. PLoS One. 2020;15(5):e0231892. doi:10.1371/journal.pone.0231892
64. Badri P, Jiang X, Borodovsky A, et al. Pharmacokinetic and pharmacodynamic properties of cemdisiran, an RNAi therapeutic targeting complement component 5, in healthy subjects and patients with paroxysmal nocturnal hemoglobinuria. Clin Pharmacokinet. 2021;60(3):365–378. doi:10.1007/s40262-020-00940-9
65. Nishimura JI, Ando K, Masuko M, et al. Tesidolumab (LFG316) for treatment of C5-variant patients with paroxysmal nocturnal hemoglobinuria. Haematologica. 2022;107(6):1483–1488. doi:10.3324/haematol.2020.265868
66. Kulasekararaj A, Brodsky R, Kulagin A, Jang JH. Biosimilars in rare diseases: a focus on paroxysmal nocturnal hemoglobinuria. Haematologica. 2023;108(5):1232–1243. doi:10.3324/haematol.2022.281562
67. Kulasekararaj A, Lanza F, Arvanitakis A, et al. Efficacy and safety of biosimilar candidate ABP 959 as compared with eculizumab reference product in paroxysmal nocturnal hemoglobinuria. Blood. 2022;140(1):8660–8662. doi:10.1182/blood-2022-166722
68. Chow V, Pan J, Chien D, Mytych DT, Hanes V. A randomized, double-blind, single-dose, three-arm, parallel group study to determine pharmacokinetic similarity of ABP 959 and eculizumab (Soliris((R))) in healthy male subjects. Eur J Haematol. 2020;105(1):66–74. doi:10.1111/ejh.13411
69. Lee HA, Jang H, Jeong D, Kim Y, Fuhr R. A randomized, double-blind, three-arm, parallel group, single-dose Phase I study to evaluate the pharmacokinetic similarity between SB12 (a proposed eculizumab biosimilar) and eculizumab (Soliris) in healthy subjects. Int J Clin Pharmacol Ther. 2022;60(6):269–279. doi:10.5414/CP204176
70. Gusarova V, Degterev M, Lyagoskin I, et al. Analytical and functional similarity of biosimilar Elizaria(R) with eculizumab reference product. J Pharm Biomed Anal. 2022;220:115004. doi:10.1016/j.jpba.2022.115004
71. Mastellos DC, Yancopoulou D, Kokkinos P, et al. Compstatin: a C3-targeted complement inhibitor reaching its prime for bedside intervention. Eur J Clin Invest. 2015;45(4):423–440. doi:10.1111/eci.12419
72. Lee JW, Brodsky RA, Nishimura JI, Kulasekararaj AG. The role of the alternative pathway in paroxysmal nocturnal hemoglobinuria and emerging treatments. Expert Rev Clin Pharmacol. 2022;15(7):851–861. doi:10.1080/17512433.2022.2109462
73. Panse J, Wilson K, Fishman J, et al. Fatigue and health-related quality of life in paroxysmal nocturnal haemoglobinuria: a post hoc analysis of the pegcetacoplan PEGASUS trial data. Eur J Haematol. 2023;111(1):72–83. doi:10.1111/ejh.13969
74. Patriquin CJ, Bogdanovic A, Griffin M, et al. Long-Term Safety and Efficacy of Pegcetacoplan Treatment in Adults with Paroxysmal Nocturnal Hemoglobinuria. Blood. 2022;140(1):2921–2923. doi:10.1182/blood-2022-163625
75. M G, Kelly R, Deeren D, et al. Intensive pegcetacoplan dosing in the management of acute hemolysis as part of the 307 open-label extension study.
76. Griffin M, Muus P, Munir T, et al. Experience of compassionate-use pegcetacoplan for paroxysmal nocturnal hemoglobinuria. Blood. 2023;141(1):116–120. doi:10.1182/blood.2022017266
77. Risitano AM, Roth A, Soret J, et al. Addition of iptacopan, an oral factor B inhibitor, to eculizumab in patients with paroxysmal nocturnal haemoglobinuria and active haemolysis: an open-label, single-arm, phase 2, proof-of-concept trial. Lancet Haematol. 2021;8(5):e344–e354. doi:10.1016/S2352-3026(21)00028-4
78. Novartis Phase III APPOINT-PNH trial shows investigational oral monotherapy iptacopan improves hemoglobin to near-normal levels, leading to transfusion Independence in all treatment-naïve PNH patients. 2023. Available from: https://www.novartis.com/news/media-releases/novartis-phase-iii-appoint-pnh-trial-shows-investigational-oral-monotherapy-iptacopan-improves-hemoglobin-near-normal-levels-leading-transfusion-independence-all-treatment-naive-pnh-patients.
79. Risitano A, Han B, Ueda Y, et al. Oral complement factor B inhibitor iptacopan monotherapy improves hemoglobin to normal/near-normal levels in paroxysmal nocturnal hemoglobinuria patients naïve to complement inhibitors: phase III APPOINT-PNH trial; 2023.
80. Barratt J, Weitz I. Complement factor D as a strategic target for regulating the alternative complement pathway. Front Immunol. 2021;12:712572. doi:10.3389/fimmu.2021.712572
81. Kulasekararaj A, Füreder W, McDonald A, et al. Factor D inhibition with oral bcx9930 leads to sustained control of hemolysis and symptoms over 48 weeks in subjects with paroxysmal nocturnal hemoglobinuria inadequately controlled on c5 inhibitors. Hemasphere. 2022;6:720–721. doi:10.1097/01.HS9.0000846188.51290.75
82. McDonald A, Malherbe JL, Kulasekararaj A, et al. Factor D inhibition with oral bcx9930 monotherapy leads to sustained control of hemolysis and symptoms over 48 weeks in subjects with paroxysmal nocturnal hemoglobinuria naïve to c5 inhibitors. Hemasphere. 2022;6:719–720. doi:10.1097/01.HS9.0000846184.35066.7a
83. BioCryst discontinues development of BCX9930 and shifts focus to potential once-daily, oral factor D inhibitor, BCX10013; 2022. Available from: https://ir.biocryst.com/news-releases/news-release-details/biocryst-discontinues-development-bcx9930-and-shifts-focus.
84. Kulasekararaj AG, Risitano AM, Maciejewski JP, et al. Phase 2 study of danicopan in patients with paroxysmal nocturnal hemoglobinuria with an inadequate response to eculizumab. Blood. 2021;138(20):1928–1938. doi:10.1182/blood.2021011388
85. Kira pharmaceuticals announces first cohort of patients dosed in phase 2 study of KP104 in paroxysmal nocturnal hemoglobinuria (PNH); 2022. Available from: https://www.prnewswire.com/news-releases/kira-pharmaceuticals-announces-first-cohort-of-patients-dosed-in-phase-2-study-of-kp104-in-paroxysmal-nocturnal-hemoglobinuria-pnh-301689797.html.
86. Griffin M, Hillmen P, Munir T, et al. Significant hemolysis is not required for thrombosis in paroxysmal nocturnal hemoglobinuria. Haematologica. 2019;104(3):e94–e96. doi:10.3324/haematol.2018.198846
87. Almeida AM, Bedrosian C, Cole A, et al. Clinical benefit of eculizumab in patients with no transfusion history in the International Paroxysmal Nocturnal Haemoglobinuria Registry. Intern Med J. 2017;47(9):1026–1034. doi:10.1111/imj.13523
88. CADTH. Ravulizumab; 2022. Available from: https://www.cadth.ca/ravulizumab-0.
89. de Castro C, Grossi F, Weitz IC, et al. C3 inhibition with pegcetacoplan in subjects with paroxysmal nocturnal hemoglobinuria treated with eculizumab. Am J Hematol. 2020;95(11):1334–1343. doi:10.1002/ajh.25960
90. Brodsky RA, Lee JW, Nishimura JI, Szer J. Lactate dehydrogenase versus haemoglobin: which one is the better marker in paroxysmal nocturnal haemoglobinuria? Br J Haematol. 2022;196(2):264–265. doi:10.1111/bjh.17860
91. Lee JW, Jang JH, Kim JS, et al. Clinical signs and symptoms associated with increased risk for thrombosis in patients with paroxysmal nocturnal hemoglobinuria from a Korean Registry. Int J Hematol. 2013;97(6):749–757. doi:10.1007/s12185-013-1346-4
92. Risitano AM, Kulasekararaj AG, Lee JW, et al. Danicopan: an oral complement factor D inhibitor for paroxysmal nocturnal hemoglobinuria. Haematologica. 2021;106(12):3188–3197. doi:10.3324/haematol.2020.261826
93. Jang JH, Pavani R, Aurand L, et al. A phase 2, randomized trial evaluating the safety and efficacy of pozelimab and cemdisiran in patients with paroxysmal nocturnal hemoglobinuria.
94. Summary Safety Review - SOLIRIS (eculizumab) and BEXSERO (Multicomponent Meningococcal B Vaccine [recombinant, adsorbed]) - Assessing the Potential Risk of Hemolysis and Low Hemoglobin in Patients Treated with Soliris and Vaccinated with Bexsero; 2016.
95. Granoff DM, Kim H, Topaz N, MacNeil J, Wang X, McNamara LA. Differential effects of therapeutic complement inhibitors on serum bactericidal activity against non-groupable meningococcal isolates recovered from patients treated with eculizumab. Haematologica. 2019;104(8):e340–e344. doi:10.3324/haematol.2018.209692
96. Konar M, Granoff DM. Eculizumab treatment and impaired opsonophagocytic killing of meningococci by whole blood from immunized adults. Blood. 2017;130(7):891–899. doi:10.1182/blood-2017-05-781450
97. Patriquin CJ, Kulasekararaj A, Peffault de Latour R, et al. Prophylactic antibiotic use and risk of meningococcal infections in patients with Paroxysmal Nocturnal Hemoglobinuria (PNH) treated with eculizumab who received meningococcal vaccination: results from the international PNH registry. Blood. 2019;134(1):4802. doi:10.1182/blood-2019-127005
98. Jodele S, Dandoy CE, Danziger-Isakov L, et al. Terminal complement blockade after hematopoietic stem cell transplantation is safe without meningococcal vaccination. Biol Blood Marrow Transplant. 2016;22(7):1337–1340. doi:10.1016/j.bbmt.2016.03.032
99. Luzzatto L, Hyry HI, Schieppati A, et al. Outrageous prices of orphan drugs: a call for collaboration. Lancet. 2018;392(10149):791–794. doi:10.1016/S0140-6736(18)31069-9
100. Kulasekararaj AG, Kuter DJ, Griffin M, Weitz IC, Roth A. Biomarkers and laboratory assessments for monitoring the treatment of patients with paroxysmal nocturnal hemoglobinuria: differences between terminal and proximal complement inhibition. Blood Rev. 2023;59:101041. doi:10.1016/j.blre.2023.101041
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.