")
Back to Journals » International Journal of General Medicine » Volume 17
Progress in the Study of the Role and Mechanism of HTRA1 in Diseases Related to Vascular Abnormalities
Authors Song S, Li X, Xue X, Dong W, Li C
Received 27 December 2023
Accepted for publication 10 April 2024
Published 18 April 2024 Volume 2024:17 Pages 1479—1491
DOI https://doi.org/10.2147/IJGM.S456912
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Redoy Ranjan
Shina Song,1,2 Xiaofeng Li,1 Xuting Xue,3 Wenping Dong,2 Changxin Li1
1Department of Neurology, The First Hospital of Shanxi Medical University, Taiyuan, People’s Republic of China; 2Department of Geriatrics, General Hospital of TISCO, Taiyuan, People’s Republic of China; 3Shanxi Key Laboratory of Otorhinolaryngology Head and Neck Cancer, The First Hospital of Shanxi Medical University, Taiyuan, People’s Republic of China
Correspondence: Wenping Dong, Department of Geriatrics, General Hospital of TISCO, North Street, Xinhualing District, Taiyuan, Shanxi Province, 030009, People’s Republic of China, Tel +86 13703581782, Email [email protected] Changxin Li, Department of Neurology, The First Hospital of Shanxi Medical University, Jiefangnan Road 85, Taiyuan, Shanxi Province, 030001, People’s Republic of China, Tel +86 13834635428, Email [email protected]
Abstract: High temperature requirement A1 (HTRA1) is a member of the serine protease family, comprising four structural domains: IGFBP domain, Kazal domain, protease domain and PDZ domain. HTRA1 encodes a serine protease, a secreted protein that is widely expressed in the vasculature. HTRA1 regulates a wide range of physiological processes through its proteolytic activity, and is also involved in a variety of vascular abnormalities-related diseases. This article reviews the role of HTRA1 in the development of vascular abnormalities-related hereditary cerebral small vessel disease (CSVD), age-related macular degeneration (AMD), tumors and other diseases. Through relevant research advances to understand the role of HTRA1 in regulating signaling pathways or refolding, translocation, degradation of extracellular matrix (ECM) proteins, thus directly or indirectly regulating angiogenesis, vascular remodeling, and playing an important role in vascular homeostasis, further understanding the mechanism of HTRA1’s role in vascular abnormality-related diseases is important for HTRA1 to be used as a therapeutic target in related diseases.
Keywords: HTRA1, vascular anomalies, cerebral small vessel disease, AMD, tumor
Introduction
High temperature requirement A1 (HTRA1) is a family of highly conserved serine proteases involved in protein quality control. This protein is widely expressed from prokaryotes to eukaryotes and exists in humans as four homologues, HTRA1, HTRA2, HTRA3 and HTRA4.1 Originally found in E. coli, HTRA1 is an important component of the heat shock response2,3 and encodes a widely expressed serine protease that is significantly expressed in the vasculature,1,4 which has both chaperone and serine protease activities and is involved in many physiological processes, particularly extracellular matrix (ECM) remodeling5–7 and transforming growth factor-β (TGF-β) signaling.1,8–11 HTRA1 plays an important role in cell proliferation, migration, and apoptosis in addition to controlling protein aggregation through refolding, translocation, or degradation.12 HTRA1 is commonly expressed in normal human tissues, but at different levels in different tissues and organs, further suggesting that HTRA1 may play different roles in different cell types. HTRA1 genes are associated with cerebral small vessel disease,13,14 age-related macular degeneration (AMD),15 multisite tumors,16–19 arthritis,20 pre-eclampsia21 and other microvascular and macrovascular diseases. The pathogenesis is mainly due to impaired vascular function.22 This review discusses the mechanisms of HTRA1’s role in the vasculature and the progress of research on related diseases. Further understanding of the direct or indirect role of HTRA1 in the vasculature may help us to develop HTRA1 as a therapeutic target.
HTRA1 Gene Abnormalities and Cerebrovascular Disease
HTRA1 and Hereditary Cerebral Small Vessel Disease (CSVD)
Cerebral small vessel disease is a common clinical cerebrovascular disease that can lead to stroke and dementia. 25% of strokes and 45% of dementia are caused by CSVD. Its harmful effects have recently received more and more attention. The clinical manifestations of cerebral small vessel disease are diverse, ranging from asymptomatic to various neurological symptoms, which can cause confusion in diagnosis and treatment.23 Cerebral Autosomal Recessive Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CARASIL) is a relatively rare hereditary small vessel disease of the brain caused by a homozygous mutation in the HTRA1 gene. The first patient with CARASIL was diagnosed in Japan in 1965, and the clinical manifestations of the disease are mainly cognitive impairment, recurrent strokes, low back pain and hair loss.24 The pathological changes observed in patients with CARASIL broadly include marked thinning of the outer membrane of small cerebral arteries, massive loss of smooth muscle cells, intimal fibrosis and thickening, and centripetal narrowing of the lumen.25–27 Dysfunction of smooth muscle cells is most prominent in small arteries.28,29
Japanese researcher Oka et al1 found that HTRA1 can bind extensively to TGF-βfamily proteins, including bone morphogenetic protein 4 (BMP4), Growth and differentiation factor 5 (Gdf5) and TGF-βs. HTRA1 was found to inhibit TGF-βfamily protein-mediated signaling. Hara30 first explored the possible molecular mechanism of CARASIL and found that HTRA1 could inhibit signaling by TGF-βfamily members, and that HTRA1 mutations led to a reduction in HTRA1 protease activity and thus failed to inhibit TGF-βsignaling, resulting in a decrease in TGF-βsignaling and increased in the intima of the affected small arteries. While the TGF-βsignaling pathway is closely linked to angiogenesis, remodeling and intravascular homeostasis,31 increased TGF-βexpression could promote the formation and accumulation of ECM (fibronectin and versican) in small intracerebral arterioles.32 This led to vascular fibrosis and dysregulation of the growth of small intracerebral blood vessels, ultimately leading to the pathogenesis of CARASIL.30
One study found that HTRA1 cleaves proTGF-β1, leading to increased TGF-β1 and TGF-β signaling-induced proteins in the cerebral small vessels of CARASIL patients, further suggesting that HTRA1 can inhibit the TGF-βsignaling pathway.10 However, contrary findings have also been made. By analysing brain tissues as well as fibroblasts from HTRA1-deficient mice and fibroblasts from CARASIL patients, it was found that HTRA1 gene mutation reduced expression of the HTRA1 protein, preventing the cleavage of latent TGF-β binding protein 1 (LTBP-1), an extracellular matrix protein that is a key regulator of TGF-β, and attenuating the TGF-βsignaling pathway transduction.11 The different results of these studies may be due to different cell types, arterial models, differences in experimental conditions or experimental methods, and different perspectives in discovering experimental problems. The pathogenic mechanism of HTRA1 gene mutation has not been fully clarified so far. In terms of molecular structure, HTRA1 protease consists of a trimer, with neighbouring HTRA1 subunits activating each other through the linker region. HTRA1 mutations may result in impaired activation cascade or inability to form a stable trimer, which may lead to impaired HTRA1 protease activity.33,34 It is currently believed that heterozygous mutations in the HTRA1 gene lead to reduced expression or loss of function of HTRA1, which in turn triggers abnormal expression of the TGFβ1/Smad signaling pathway, ultimately leading to the expression of downstream signaling molecules and target proteins in heterozygous HTRA1 mutations.
Cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) can be divided into 2 types, one is CADASIL type 1 (CADASIL1, OMIM 125310), caused by mutations in the NOTCH receptor 3 gene (NOTCH3), is the most common hereditary cerebral small vessel disease35 and is broadly referred to collectively as CADASIL, while the other type, CADASIL type 2 (CADASIL2, OMIM 616779), also known as symptomatic carriers of HTRA1 gene mutations,33,36 and studies have found that HTRA1 heterozygous mutations can cause cerebrovascular abnormalities.37 The clinical manifestations of CARASIL and symptomatic carriers of HTRA1 mutations are approximately the same, but the latter have milder clinical symptoms14,38 and later onset.33,39,40 MRI of the head showed typical features of cerebral small-vessel disease, with pathological manifestations of intimal thickening, epicardial fibrosis, degeneration and loss of smooth muscle cells, and delamination and splitting of the internal elastic lamina of small vessels.37 There were no osmiophilic granules on the surface of vascular smooth muscle on electron microscopy. It has been shown that mutations in the heterozygous HTRA1 gene result in a reduction in the protein hydrolysis activity of HTRA1,41 which is accompanied by an increase in the levels of TGF-β1/Smad proteins, with a similar molecular mechanism to CARASIL.42
Although CARASIL and CADASIL in the broad sense are two different diseases caused by mutations, there is a close association between HTRA1 and NOTCH, and HTRA1 is directly related to NOTCH3 signaling regulation.28 CADASIL is caused by mutations in the NOTCH3 gene resulting in excessive accumulation of the NOTCH3 extracellular structural domain (NOTCH3-ECD) and vascular wall disruption.43 Zellner et al44 quantified the cerebrovascular proteome from CADASIL patients and control autopsy samples and obtained 95 proteins with significantly increased abundance and found that HTRA1 was highly enriched and colocalized with NOTCH3-ECD deposition in the patient’s vasculature, which provides a basis for the development of CADASIL pathology during provides a key step in connecting the molecular mechanisms of two distinct cerebral small vessel diseases.
In HTRA1 associated cerebral small-vessel disease, researchers have focused more on the vascular smooth muscle cell (VSMC), and disturbances in VSMC function and integrity were the main causative agents of hereditary CSVD, which may lead to impaired blood perfusion in certain regions of the brain.45 Further mechanistic studies on the phenotypic transformation of VSMCs by Klose et al28 found that HTRA1 deletion increased NOTCH3 ligand JAG1 protein levels and NOTCH3 signaling activity in VSMCs, with activation of NOTCH3 signaling leading to increased transcription of HES and HEY repressors and contributing to the contractile phenotype of VSMC. The contractile phenotype VSMCs play an important role in the control of blood flow and blood pressure. Contractile phenotype VSMCs are characterised by slow proliferation and association with neurotransmitters.46 The VSMC synthetic phenotype cell morphology is spindle-shaped and the main features are proliferation, migration and production of extracellular matrix.46 In VSMC, HES and HEY proteins can inhibit transcription of VSMC contraction phenotype marker proteins such as SM22α, α-SMA, and Smoothelin expression, causing VMSC to turn into a synthetic phenotype, resulting in aberrant VMSC function.47 JAG1 and NOTCH3 are the most abundantly expressed ligands and receptors on VSMC.48,49 NOTCH3-ICD activates the transcription of contractile proteins, whereas Notch target genes of the HES and HEY families encode transcriptional repressor proteins that inhibit the expression of these contractile VSMC markers28 (Figure 1). Cerebral arterial vascular abnormalities were observed in HTRA1-/- mice, from which aortic VMSCs were isolated that were predominantly of the synthetic phenotype and showed enhanced proliferation and migration.29 The synthetic phenotype of VMSCs induces elevated expression of matrix metalloproteinases (MMPs), which degrade the extracellular matrix and may lead to the increased VSMC death. Abnormal synthetic regulation may be one of the mechanisms by which HTRA1 deficiency causes vascular abnormalities29 (Figure 1).
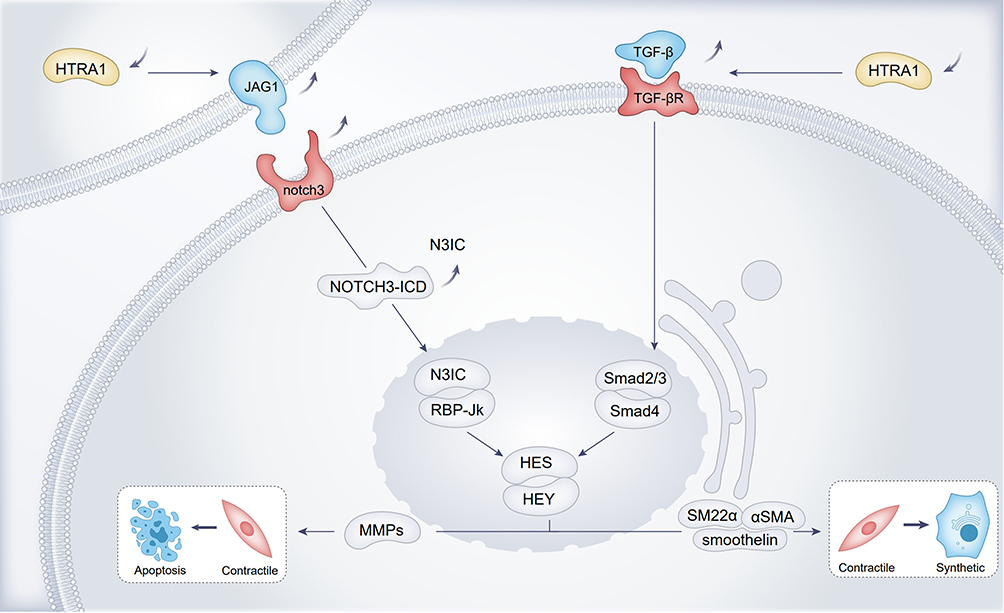 |
Figure 1 Loss of function of HTRA1 induces an increased VMSC synthetic phenotype and VMSC apoptosis. |
Kato et al50 found that HTRA1-/- mice exhibited pathological features of CARASIL-associated arteriopathy, and that reduced HTRA1 expression could lead to the accumulation of matrisome proteins such as hub protein fibronectin (FN) and latent TGF-β binding protein 4 (LTBP-4). Candesartan treatment, however, attenuated matrisome proteins accumulation and normalised vasodilation and cerebral blood flow. Candesartan, an inhibitor of TGF-β signaling,51–53 amend arteriopathy in HTRA1-/- mice, thus suggesting that drugs with HTRA1 activity may be new therapeutic candidates for CARASIL, and that these approaches may open new pathways for preventing the progression of CSVD.
In summary, in HTRA1-associated cerebral small vessel disease, downregulation of HTRA1 protein expression can affect TGF-β signaling pathway transduction, which indirectly regulates angiogenesis, influences the phenotypic changes of VSMCs in the vasculature, and induces an increase in the expression of MMP9, which leads to the death of VMSCs. Meanwhile, FN, LTBP-1, LTBP-4 and other related matrisome proteins were increased in the vascular endothelium, leading to changes in vascular structure. In conclusion, reduced HTRA1 protease activity can lead to growth dysregulation of small blood vessels in the brain, and more studies are needed to differentiate the pathology, clinical features, and mechanisms of action between CARASIL and heterozygous mutation carriers.
HTRA1 and Neurodegenerative Lesions, Cerebral Microhemorrhages and Cerebral Amyloidosis
HTRA1 is associated with neurodegenerative lesions, cerebral microhemorrhages, cerebral amyloidosis, and in a Finnish cohort study, investigators performed whole-exome sequencing (WES) in patients with cerebrovascular cognitive impairment (VCI), supporting a pathogenic role for HTRA1 in cerebrovascular cognitive impairment and cerebral small vessel disease, considering vasculogenic mechanisms associated with neurodegenerative diseases54,55 and providing new insights into the molecular mechanisms of cerebrovascular cognitive disorders diseases.
Mild to moderate cerebral microhemorrhages are common in CARASIL patients, but diffuse cerebral microhemorrhages are rare. Wen et al56 reported a 43-year-old woman whose imaging showed multiple cerebral microhemorrhages in all lobes, the brainstem and the cerebellum. After sequencing, it was found to be a novel mutation in HTRA1, so HTRA1 gene mutation was considered to be associated with cerebral microbleeds.
Cerebral amyloid angiopathy (CAA) is an age-related disease that is a major cause of cerebral hemorrhage and cognitive decline. In CAA patients, amyloid-β (Aβ) deposits in the vasculature and leads to disruption of the structural integrity of the vasculature, resulting in the development of the disease.57 Various fragments of amyloid precursor proteins, involved in the amyloid pathway, are co-localized with Aβ deposition in human brain samples.58 Zellner et al using proteomics techniques, also similarly found that HTRA1 co-localized with Aβ deposition in brain capillaries of CAA type 1 patient’s vascular. The upregulation of HTRA1 levels may reflect extracellular matrix remodeling in the vasculature as a compensatory response to vascular Aβ accumulation.59 These findings suggest that abnormal HTRA1-mediated protein degradation plays an important role in CAA type 1 microvasculature and has a molecular link between multiple types of cerebral microvascular disease.60
HTRA1 is Associated with Age-Related Macular Degeneration (AMD)
Age-related macular degeneration is a chronic progressive disease characterized by chronic progressive degeneration of the macular photoreceptors, retinal pigment epithelium, bruch’s membrane (BrM) and choroidal capillaries.61 In the clinic, age-related macular degeneration initially causes damage to the central area of the retina (the macula) and is divided into early and late stages (advanced age-related macular degeneration). The advanced stages of AMD are divided into non-exudative or atrophic AMD (dry AMD) and exudative or neovascularised AMD (wet AMD).62 In dry AMD, drusen deposits are observed between the retinal pigment epithelium and the BrM membrane with progressive development of geographic atrophy, whereas in wet AMD, vision loss occurs due to macular choroidal neovascularization (CNV) or polypoidal choroidal vasculopathy (PCV).63 PCV and CNV are considered as subtypes of wet AMD. Patients with wet AMD have more rapid vision loss than those with dry AMD.
Most studies have shown that HTRA1 gene variants are significantly associated with wet AMD disease.64–68 The HTRA1 single nucleotide polymorphism (SNP) locus rs11200638, located on human chromosome 10q26, was significantly associated with AMD. This SNP increased the expression of HTRA1 protein in the retinal pigment epithelium of AMD patients.69 It was shown that HTRA1 rs11200638 has a stronger pathogenic risk for wet AMD than for dry AMD.70 Because of the clear correlation between wet AMD and vascularity, here we focus on wet AMD. The results of the exploration of the role of HTRA1 in the pathogenesis of PCV can help guide the corresponding clinical treatment.
HTRA1 and Wet AMD
HTRA1 overexpression is associated with choroidal neovascularisation in the retina and macula.12,71,72 The main manifestations include abnormal enlargement of choroidal vessels, loss of the elastic lamina inside as well as outside the choroidal arteries, with severe regression or absence of the middle layer.73–75 In a recent study, HTRA1 modulated subclinical inflammation of retina and choroid, and it has been confirmed by in vivo and in vitro experiments that HTRA1 had a pro-angiogenic function, and the inflammatory environment induced by HTRA1 overexpression may increase angiogenic response.76
HTRA1 acts on many key components of the BrM, and it hydrolyses ECM such as aggrecan, decorin, type II collagen,77 fibronectin, fibromodulin, and vitronectin,5 as well as being involved in degradation of elastin fibres in the choroidal vasculature and the BrM membrane.78 Degradation of proteins in the BrM membrane makes it easier for blood vessels to grow from the choroid across the BrM membrane to the neural retina, leading to the development of AMD. Notably, the ECM fragments produced by HTRA1 hydrolysis of these ECMs may themselves promote angiogenesis, further exacerbating choroidal and RPE disruption.79 Transgenic mice overexpressing equal amounts of human HTRA1 in the RPE were found to have elevated expression of fibronectin fragments in the REP and choroidal layers and reduced levels of fibulin-5 and tropoelastin protein expression.7 Fibulin-5 can bind to tropoelastin, promote elastin maturation, accelerate elastic fibre synthesis, and play a role in promoting fibre synthesis.80 At the same time, the researchers found that the BrM membranes of this mouse were fragmented and the continuity was deteriorated. (Figure 2)
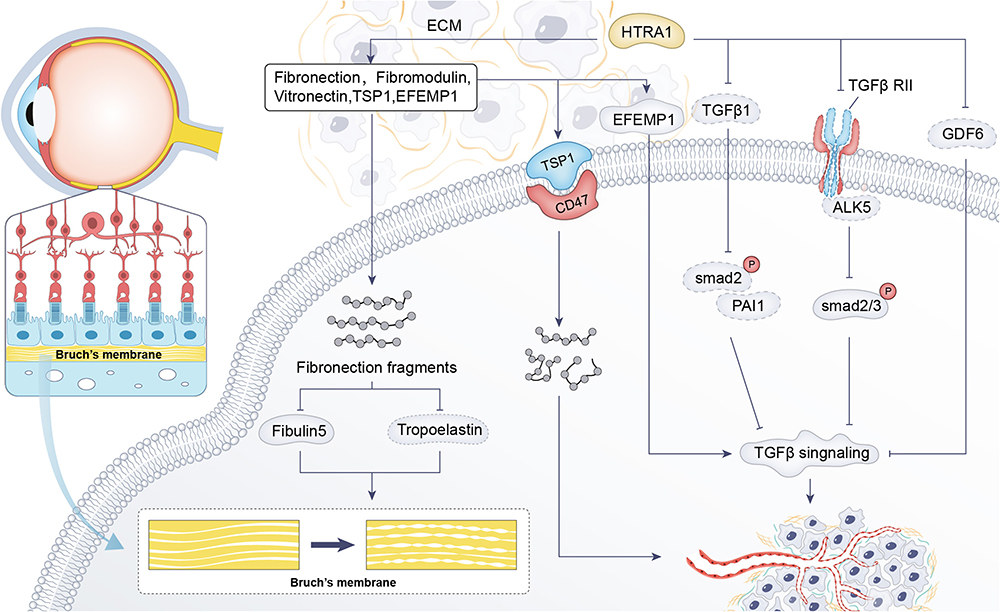 |
Figure 2 HTRA1 can affect BrM morphology through cleavage of ECM (Fibronection). HTRA1 affects angiogenesis through ECM (TSP1, EFEMP1) and TGF-β signaling pathways (TGF-β, TGF-βRII, GDF6)-related proteins and receptors in AMD. |
In induced pluripotent stem (iPS)-RPE cells at high risk for the ARMS2/HTRA1 allele, expression of the HTRA1 substrate EFEMP1 is elevated. EFEMP1 is an extracellular matrix protein that causes drusen deposition, activates the complement pathway, induces an inflammatory response, and regulates the TGF-β signaling pathway thereby affecting angiogenesis.81 Thrombospondin (TSP1) was also found to be elevated.81,82 The possible reason for the increase in ECMs was that HTRA1 cleavage activity is elevated to a feedback loop that produces more HTRA1 cleavage substrates. TSP1 is a well known inhibitor of angiogenesis and mediates differential effects through interaction with CD47.83,84 Although TSP1 itself is anti-neovascular, hydrolysis produces fragments that promote endothelial cell lumen formation and hence neovascularization.81 (Figure 2)
HTRA1 interacts with a variety of receptor-binding factors of the TGF-β family to mediate the regulation of angiogenesis, of which growth differentiation factor 6 (GDF6) is a member of the TGF-β family.85 Mutations in the GDF6 gene are associated with phenotypes such as microphthalmia and anophthalmia, which control the development of blood vessels and nerves in the eyes.86 The interaction between GDF6 and HTRA1 is crucial for TGF-β signal transduction and ocular vascular and nerve development. HTRA1-associated PCV risk genes (rs10490924T, rs6982567) were associated with elevated HTRA1 levels and reduced GDF6 levels, leading to an increased risk of AMD.85–87 In addition, AMD-associated synonymous variants in HTRA1 downregulate the expression of TGF-β1 downstream proteins such as phosphorylated SMAD2 and PAI-1, which leads to impaired TGF-β signaling regulation, thus further affecting vascular and neuromodulation88 (Figure 2). HTRA1 inhibits transforming growth factor-β2 (TGF-β2) signaling, an important regulator of angiogenesis.89 TGF-β2 knockout mice exhibit abnormal ocular morphogenesis phenotypes in the retina.90 Because of the important role of TGF-β2/activin receptor-like kinase 5 (ALK5) / SMAD2/3 signaling in neovascularisation,91 researchers92 used the CAG-promoter transgenic mice overexpressing HtrA1, found that HTRA1 cleaves TGF-β RII and inhibits SMAD2/3 phosphorylation, resulting in reduced protein expression of the downstream TGF-β- ALK5 -SMAD2/3 signaling pathway (Figure 2).
In vivo and in vitro experiments, pathology and genetics have helped to provide a deeper understanding of the pathogenesis of AMD. Structurally, HTRA1 disrupts the integrity of the choroidal vasculature and Bruch’s membrane; genetically, the HTRA1 locus has been shown to be one of the risk loci most strongly associated with wet AMD; Molecularly and biologically, HTRA1 can regulate ocular neovasculogenesis through the effects of cleavage of ECM as well as modulation of signaling pathways, which are involved in the pathogenesis of wet AMD.
HTRA1 and VEGF-Related Content
Wet AMD is characterized by vascular lesions leading to plasmacytic exudation and hemorrhage, and studies have identified an association with abnormal angiogenesis mediated by vascular endothelial growth factor (VEGF).93,94 VEGF is a highly conserved homodimeric glycoprotein that regulates physiological and pathological angiogenesis. It is also a potent vascular permeability factor, increasing the permeability of the vessel wall.95 Studies have confirmed that VEGF can induce neovascularization and that anti-VEGF treatment can significantly improve patients’ visual acuity.96,97 A meta-analysis showed a significant association between the HTRA1rs11200638 gene and response to anti-VEGF treatment, and blocking the angiogenic pathway may be partly responsible for the effect of HRTA1 on response to anti-VEGF treatment.98 In vitro and in vivo experimental studies have shown that HTRA1 can synergize with oxidized phospholipids to promote inflammation and macrophage infiltration, which are essential for ocular VEGF expression, and that HTRA1 may mediate VEGF expression in RPE cells through the Wnt signaling pathway, further suggesting that, inhibition of HTRA1 expression reduces choroidal neovascularization in mouse disease models.99
Binding of the Gtf2i-β/δ transcription factor to the ARMS2 gene leads to elevated HTRA1-secreting proteins in transfected cells and AMD-derived induced pluripotent stem cells (iPSCs), overexpression of HTRA1 in mice using the CAG-promoter, and increased blood levels of HTRA1 protein, causing VEGF upregulation and a CNV-like phenotype.92 Tom et al75 developed a Fab fragment to block HTRA1, using Dick-kopf-related protein 3 (DKK3) as a biomarker, and demonstrated that in AMD patients treated with an HTRA1-blocking Fab fragment, HTRA1 activity was inhibited in AMD patients treated with HTRA1 blocking Fab fragment. Thus the anti-HTRA1 approach offers a promising alternative treatment option for wet AMD that is complementary to anti-VEGF therapy.
Research Progress of HTRA1 Gene Abnormalities Associated with Tumors
Tumor angiogenesis is closely related to tumor invasion, infiltration, and metastasis, which directly affects the prognosis of tumor patients, and HTRA1 may be involved in tumorigenesis and tumor angiogenesis. The expression of HTRA1 was found to be downregulated in most tumors.100 Overexpression of HTRA1 in highly aggressive melanoma, oesophageal squamous cell carcinoma and pancreatic cancer cells inhibits the proliferation and migration of tumor cells and also causes apoptosis.18,101,102 HTRA1 can promote cancer cell death through apoptosis and oxidative stress pathways.103,104 Another study reported that HTRA1 overexpression in ovarian cancer cells inhibited epidermal growth factor receptor (EGFR) activation, leading to a decrease in AKT/MAPK phosphorylation levels and a significant increase in cell death.105 Certain anticancer drugs, such as cisplatin and paclitaxel, can upregulate HTRA1, which was found to initiate programmed cell death through autocatalytic activation.104 Thus HTRA1 is considered to be a tumor suppressor gene in some cancers that promotes cell death. However, it has also been shown that HTRA1 is an oncogene that promotes tumor cell metastasis.106,107
The current studies on HTRA1 in tumor vascular related mechanisms of action are as follows, in terms of tumor growth inhibition, Klose et al22 investigated the effect of HTRA1 on tumor angiogenesis. NOTCH1 signaling regulates endothelial cell proliferation, migration and tube formation. Experiments further confirmed that HTRA1 plays a key role in regulating NOTCH1 signaling by cleaving JAG1.22 JAG1 antagonises the NOTCH ligand DLL4 (Notch ligand Delta-like 4), which has anti-angiogenic effects.108 Inhibition of NOTCH1 signaling can lead to increased VEGFR2 expression, which further affects angiogenesis.109 Moreover, inhibition of the DLL4 can also lead to elevated VEGFR2 expression.110 This ultimately leads to abnormal functioning of the vascular network as well as increased hypoxia in the tumor tissue, thereby inhibiting tumor growth (Figure 3).
 |
Figure 3 Mechanistic studies of HTRA1 associated with the tumor microenvironment. (A) PN-1 expression is regulated through the EGF /PKC-δ/MEK/ERK/EGR1 signaling pathway to inhibit the effect of the HTRA1 protein on tumors; (B) HTRA1 promotes the transdifferentiation of normal fibroblasts to CAF by activating the bFGF/FGF2 signaling axis secreted by gastric cancer cells through activation of the NF-kB signaling pathway; (C) in vascular endothelial cells, HTRA1 affects angiogenesis through the NOTCH signaling pathway. |
With regard to the promotion of tumor growth by HTRA1, the main focus has been on the effects on the tumor stroma, the main component of which is the tumor vasculature. HTRA1 can promote tumor growth by inducing cancer-associated fibroblasts (CAF).111 CAFs secrete a range of cytokines to regulate the tumor vascular network, and CAFs in the tumor microenvironment contribute to tumourigenesis.111 α-SMA is a surface marker for CAFs.107 Wu et al19 found that HTRA1 was positively correlated with α-SMA expression in gastric cancer tissues, and HTRA1 promoted the bFGF/FGF2 signaling axis secreted by gastric cancer cells through activation of the NF-kB signaling pathway, which in turn facilitated the transdifferentiation of normal fibroblasts to CAFs (Figure 3). Knockdown of HTRA1 inhibits migration and invasion of breast epithelial cells in vitro.112 In the tumor microenvironment, Protease nexin-1 (PN-1) could increase breast cancer cell migration and invasion through the epidermal growth factor (EGF) / protein kinase (PKC)/ Mitogen-activated protein kinase (MAPK)/ early growth response protein 1 (EGR1) axis.113 PN-1 promoted cancer cell metastasis mainly by remodeling the tumor stroma. EGF expression was elevated in the breast tumor microenvironment, which upregulated the expression of PN-1 by binding to epidermal growth factor receptor (EGFR), activating the downstream protein kinase C-δ (PKC-δ), extracellular signal-regulated kinase (ERK), MEK and its transcription factor EGR1. PN-1 blocks the function of HtrA1, which disrupts the cleavage of EGF, leading to further activation of EGF signaling, acting as a feedback signal to increase PN-1 expression113 (Figure 3).
In addition, it has been shown that HTRA1 bind to the tumor promoter lysyl oxidase (LOX), which inhibits TGF-β1 signaling pathway transduction and increases the expression of Matrilin2 (MATN2), further increasing the expression of the EGFR to promote tumor progression and metastasis.114 TGF-β is associated with HTRA1 during tumorigenesis via the Wnt/β-catenin signaling pathway, EMT and EGFR.105,115–117 In the early stages of cancer, the TGF-β signaling pathway can act as a tumor suppressor to induce cell death.118 In advanced stages of cancer, the TGF-β signaling pathway can act as a tumor promoter in the tumor microenvironment.119 Further studies are needed to unravel the complex interactions between HTRA1 and TGF-β in a cell type-dependent manner, which is necessary to understand HTRA1 as a multifunctional regulator in cancer. Therefore, in the tumor mesenchyme, further investigation of the effects of HTRA1 on the tumor mesenchyme, especially the tumor vasculature, could enhance our understanding of the role of HTRA1 in normal and malignant cells.
Problems and Prospects
The molecular mechanism of human HTRA1 action is a promising study, and current research on HTRA1 is still in its early stages, with increasing evidence that HTRA1 plays a role in the regulation of development, cell growth, differentiation, and apoptosis. HTRA1 is associated with diseases such as cerebrovascular disease, AMD and tumors, mainly in terms of vascular changes in the associated diseases, suggesting that whether it is the endothelial cells, smooth muscle cells, fibroblasts, and other structures related to the vasculature of the blood vessels, HTRA1 has an impact on their functions and physiological processes. HTRA1 plays an important role in vascular homeostasis by regulating signaling pathways or refolding, translocation, and degradation of extracellular matrix proteins, thereby directly or indirectly regulating angiogenesis and vascular remodeling, and vascular and related structural dysfunction caused by abnormal function of the HTRA1 gene is an important initiator and high risk factor. This article suggests that more research on vascular diseases involving cardiovascular,120 renal,121 and hepatic diseases122 should include HTRA1 as an important research target, and even in the process of tissue repair and regeneration,123 scarring,124 senescence,125,126 and pregnancy complications87 should include HTRA1 as an indispensable research component.
To fully understand the role of HTRA1 in disease, in particular, further study of the mechanisms of HTRA1 action in the vasculature could provide a theoretical basis for clinical practice, which is essential to understand their relevance in cellular physiology and pathogenesis and their use as targets in the future treatment of diseases. Future indepth studies on HTRA1 and vascular-related signaling pathway transduction are believed to lead to more discoveries about HTRA1 and provide new ideas for clinical diagnosis and treatment.
Data Sharing Statement
All data supporting the findings of this study appear in the submitted manuscript or are available from the corresponding author upon reasonable request.
Funding
This research was funded by Shanxi Scientific and Technological Innovation Programs of Higher Education Institutions (2023L105) and Shanxi Province Basic Research Project (202103021224404).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Oka C, Tsujimoto R, Kajikawa M, et al. HtrA1 serine protease inhibits signaling mediated by Tgfβ family proteins. Development. 2004;131(5):1041–1053. doi:10.1242/dev.00999
2. Zumbrunn J, Trueb B. Primary structure of a putative serine protease specific for IGF-binding proteins. FEBS Lett. 1996;398(2–3):187–192. doi:10.1016/S0014-5793(96)01229-X
3. Hu SI, Carozza M, Klein M, Nantermet P, Luk D, Crowl RM. Human HtrA, an evolutionarily conserved serine protease identified as a differentially expressed gene product in osteoarthritic cartilage. J Biol Chem. 1998;273(51):34406–34412. doi:10.1074/jbc.273.51.34406
4. De Luca A, De Falco M, Severino A, et al. Distribution of the serine protease HtrA1 in normal human tissues. J Histochem Cytochem. 2003;51(10):1279–1284.
5. An E, Sen S, Park SK, Gordish-Dressman H, Hathout Y. Identification of novel substrates for the serine protease HTRA1 in the human RPE secretome. Invest Ophthalmol Vis Sci. 2010;51(7):3379–3386. doi:10.1167/iovs.09-4853
6. Tiaden AN, Breiden M, Mirsaidi A, et al. Human serine protease HTRA1 positively regulates osteogenesis of human bone marrow-derived mesenchymal stem cells and mineralization of differentiating bone-forming cells through the modulation of extracellular matrix protein. Stem Cells. 2012;30(10):2271–2282. doi:10.1002/stem.1190
7. Vierkotten S, Muether PS, Fauser S, Lewin A. Overexpression of HTRA1 leads to ultrastructural changes in the elastic layer of Bruch’s membrane via cleavage of extracellular matrix components. PLoS One. 2011;6(8):e22959. doi:10.1371/journal.pone.0022959
8. Graham JR, Chamberland A, Lin Q, et al. Serine protease HTRA1 antagonizes transforming growth factor-beta signaling by cleaving its receptors and loss of HTRA1 in vivo enhances bone formation. PLoS One. 2013;8(9):e74094. doi:10.1371/journal.pone.0074094
9. Launay S, Maubert E, Lebeurrier N, et al. HtrA1-dependent proteolysis of TGF-beta controls both neuronal maturation and developmental survival. Cell Death Differ. 2008;15(9):1408–1416. doi:10.1038/cdd.2008.82
10. Shiga A, Nozaki H, Yokoseki A, et al. Cerebral small-vessel disease protein HTRA1 controls the amount of TGF-beta1 via cleavage of proTGF-beta1. Hum Mol Genet. 2011;20(9):1800–1810. doi:10.1093/hmg/ddr063
11. Beaufort N, Scharrer E, Kremmer E, et al. Cerebral small vessel disease-related protease HtrA1 processes latent TGF-beta binding protein 1 and facilitates TGF-beta signaling. Proc Natl Acad Sci U S A. 2014;111(46):16496–16501. doi:10.1073/pnas.1418087111
12. Oka C, Saleh R, Bessho Y, Reza HM. Interplay between HTRA1 and classical signalling pathways in organogenesis and diseases. Saudi J Biol Sci. 2022;29(4):1919–1927. doi:10.1016/j.sjbs.2021.11.056
13. Uemura M, Nozaki H, Kato T, et al. HTRA1-related cerebral small vessel disease: a review of the literature. Front Neurol. 2020;11:545. doi:10.3389/fneur.2020.00545
14. Liu JY, Zhu YC, Zhou LX, et al. HTRA1-related autosomal dominant cerebral small vessel disease. Chin Med J. 2020;134(2):178–184. doi:10.1097/CM9.0000000000001176
15. Liao SM, Zheng W, Zhu J, et al. Specific correlation between the major chromosome 10q26 haplotype conferring risk for age-related macular degeneration and the expression of HTRA1. Mol Vis. 2017;23:318–333.
16. Li Y, Yuan J, Rothzerg E, et al. Molecular structure and the role of high-temperature requirement protein 1 in skeletal disorders and cancers. Cell Prolif. 2020;53(2):e12746. doi:10.1111/cpr.12746
17. Yao Y, Li N. Effect of HtrA1 polymorphism on sensitivity to chemotherapy in patients with colon cancer. Med Sci Monit. 2020;26:e921933. doi:10.12659/MSM.921933
18. Cheng H, Zhu H, Cao M, Lu C, Bao S, Pan Y. HtrA1 suppresses the growth of pancreatic cancer cells by modulating Notch-1 expression. Braz J Med Biol Res. 2018;52(1):e7718. doi:10.1590/1414-431x20187718
19. Wu H, Ma S, Xiang M, Tong S. HTRA1 promotes transdifferentiation of normal fibroblasts to cancer-associated fibroblasts through activation of the NF-kappaB/bFGF signaling pathway in gastric cancer. Biochem Biophys Res Commun. 2019;514(3):933–939. doi:10.1016/j.bbrc.2019.05.076
20. Tiaden AN, Richards PJ. The emerging roles of HTRA1 in musculoskeletal disease. Am J Pathol. 2013;182(5):1482–1488. doi:10.1016/j.ajpath.2013.02.003
21. Teoh SS, Zhao M, Wang Y, Chen Q, Nie G. Serum HtrA1 is differentially regulated between early-onset and late-onset preeclampsia. Placenta. 2015;36(9):990–995. doi:10.1016/j.placenta.2015.07.001
22. Klose R, Adam MG, Weis EM, et al. Inactivation of the serine protease HTRA1 inhibits tumor growth by deregulating angiogenesis. Oncogene. 2018;37(31):4260–4272. doi:10.1038/s41388-018-0258-4
23. Markus HS, van Der Flier WM, Smith EE, et al. Framework for Clinical Trials in Cerebral Small Vessel Disease (FINESSE): a review. JAMA Neurol. 2022;79(11):1187–1198. doi:10.1001/jamaneurol.2022.2262
24. Fukutake T. Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL): from discovery to gene identification. J Stroke Cerebrovasc Dis. 2011;20(2):85–93. doi:10.1016/j.jstrokecerebrovasdis.2010.11.008
25. Arima K, Yanagawa S, Ito N, Ikeda S. Cerebral arterial pathology of CADASIL and CARASIL (Maeda syndrome). Neuropathology. 2003;23(4):327–334. doi:10.1046/j.1440-1789.2003.00519.x
26. Yu Z, Cao S, Wu A, et al. Genetically confirmed CARASIL: case report with novel HTRA1 mutation and literature review. World Neurosurg. 2020;143:121–128. doi:10.1016/j.wneu.2020.05.128
27. Ito J, Nozaki H, Toyoshima Y, et al. Histopathologic features of an autopsied patient with cerebral small vessel disease and a heterozygous HTRA1 mutation. Neuropathology. 2018;38(4):428–432. doi:10.1111/neup.12473
28. Klose R, Prinz A, Tetzlaff F, et al. Loss of the serine protease HTRA1 impairs smooth muscle cells maturation. Sci Rep. 2019;9(1):18224. doi:10.1038/s41598-019-54807-6
29. Ikawati M, Kawaichi M, Oka C, Bader M. Loss of HtrA1 serine protease induces synthetic modulation of aortic vascular smooth muscle cells. PLoS One. 2018;13(5):e0196628. doi:10.1371/journal.pone.0196628
30. Hara K, Shiga A, Fukutake T, et al. Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N Engl J Med. 2009;360(17):1729–1739. doi:10.1056/NEJMoa0801560
31. Hachana S, Larrivée B. TGF-β superfamily signaling in the eye: implications for ocular pathologies. Cells. 2022;11(15):2336. doi:10.3390/cells11152336
32. Shiga A, Nozaki H, Nishizawa M, Onodera O. 常染色体劣性脳動脈症の分子病態 [Molecular pathogenesis of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy]. Brain Nerve. 2010;62(6):595–599. Japanese.
33. Verdura E, Herve D, Scharrer E, et al. Heterozygous HTRA1 mutations are associated with autosomal dominant cerebral small vessel disease. Brain. 2015;138(Pt 8):2347–2358. doi:10.1093/brain/awv155
34. Nozaki H, Kato T, Nihonmatsu M, et al. Distinct molecular mechanisms of HTRA1 mutants in manifesting heterozygotes with CARASIL. Neurology. 2016;86(21):1964–1974. doi:10.1212/WNL.0000000000002694
35. Opherk C, Gonik M, Duering M, et al. Genome-wide genotyping demonstrates a polygenic risk score associated with white matter hyperintensity volume in CADASIL. Stroke. 2014;45(4):968–972.
36. Chen X, Deng S, Xu H, et al. Novel and recurring NOTCH3 mutations in two Chinese patients with CADASIL. Neurodegener Dis. 2019;19(1):35–42. doi:10.1159/000500166
37. Cao H, Liu J, Tian W, et al. A novel heterozygous HTRA1 mutation in an Asian family with CADASIL-like disease. J Clin Lab Anal. 2022;36(2):e24174. doi:10.1002/jcla.24174
38. Bekircan-Kurt CE, Cetinkaya A, Gocmen R, et al. One disease with two faces: semidominant inheritance of a novel HTRA1 mutation in a consanguineous family. J Stroke Cerebrovasc Dis. 2021;30(9):105997. doi:10.1016/j.jstrokecerebrovasdis.2021.105997
39. Rutten JW, Haan J, Terwindt GM, van Duinen SG, Boon EM, Lesnik Oberstein SA. Interpretation of NOTCH3 mutations in the diagnosis of CADASIL. Expert Rev Mol Diagn. 2014;14(5):593–603. doi:10.1586/14737159.2014.922880
40. Di Donato I, Bianchi S, Gallus GN, et al. Heterozygous mutations of HTRA1 gene in patients with familial cerebral small vessel disease. CNS Neurosci Ther. 2017;23(9):759–765. doi:10.1111/cns.12722
41. Fasano A, Formichi P, Taglia I, et al. HTRA1 expression profile and activity on TGF-beta signaling in HTRA1 mutation carriers. J Cell Physiol. 2020;235(10):7120–7127. doi:10.1002/jcp.29609
42. Zhuo ZL, Cong L, Zhang J, Zhao XT. A novel heterozygous HTRA1 mutation is associated with autosomal dominant hereditary cerebral small vessel disease. Mol Genet Genomic Med. 2020;8(6):e1111. doi:10.1002/mgg3.1111
43. Oliveira DV, Coupland KG, Shao W, et al. Active immunotherapy reduces NOTCH3 deposition in brain capillaries in a CADASIL mouse model. EMBO Mol Med. 2023;15(2):e16556. doi:10.15252/emmm.202216556
44. Zellner A, Scharrer E, Arzberger T, et al. CADASIL brain vessels show a HTRA1 loss-of-function profile. Acta Neuropathol. 2018;136(1):111–125. doi:10.1007/s00401-018-1853-8
45. Tan RY, Markus HS. Monogenic causes of stroke: now and the future. J Neurol. 2015;262(12):2601–2616. doi:10.1007/s00415-015-7794-4
46. Bundy K, Boone J, Simpson CL. Wnt signaling in vascular calcification. Front Cardiovasc Med. 2021;8:708470. doi:10.3389/fcvm.2021.708470
47. Morris HE, Neves KB, Montezano AC, MacLean MR, Touyz RM. Notch3 signalling and vascular remodelling in pulmonary arterial hypertension. Clin Sci. 2019;133(24):2481–2498. doi:10.1042/CS20190835
48. Boucher J, Gridley T, Liaw L. Molecular pathways of notch signaling in vascular smooth muscle cells. Front Physiol. 2012;3:81. doi:10.3389/fphys.2012.00081
49. Fouillade C, Monet-Lepretre M, Baron-Menguy C, Joutel A. Notch signalling in smooth muscle cells during development and disease. Cardiovasc Res. 2012;95(2):138–146. doi:10.1093/cvr/cvs019
50. Kato T, Manabe RI, Igarashi H, et al. Candesartan prevents arteriopathy progression in cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy model. J Clin Invest. 2021;131(22). doi:10.1172/JCI140555
51. Lanz TV, Ding Z, Ho PP, et al. Angiotensin II sustains brain inflammation in mice via TGF-beta. J Clin Invest. 2010;120(8):2782–2794. doi:10.1172/JCI41709
52. Bar-Klein G, Cacheaux LP, Kamintsky L, et al. Losartan prevents acquired epilepsy via TGF-beta signaling suppression. Ann Neurol. 2014;75(6):864–875. doi:10.1002/ana.24147
53. Ongali B, Nicolakakis N, Tong XK, Lecrux C, Imboden H, Hamel E. Transforming growth factor-β1 (TGF-β1) induces cerebrovascular dysfunction and astrogliosis through angiotensin II type 1 receptor-mediated signaling pathways. Can J Physiol Pharmacol. 2018;96(5):527–534. doi:10.1139/cjpp-2017-0640
54. Monkare S, Kuuluvainen L, Schleutker J, et al. Genetic analysis reveals novel variants for vascular cognitive impairment. Acta Neurol Scand. 2022;146(1):42–50. doi:10.1111/ane.13613
55. Monkare S, Kuuluvainen L, Kun-Rodrigues C, et al. Whole-exome sequencing of Finnish patients with vascular cognitive impairment. Eur J Hum Genet. 2021;29(4):663–671. doi:10.1038/s41431-020-00775-9
56. Wen L, Yuan J, Li S, et al. Case report: diffuse cerebral microbleeds in cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy. Front Neurol. 2022;13:818332. doi:10.3389/fneur.2022.818332
57. Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease - one peptide, two pathways. Nat Rev Neurol. 2020;16(1):30–42. doi:10.1038/s41582-019-0281-2
58. Grau S, Baldi A, Bussani R, et al. Implications of the serine protease HtrA1 in amyloid precursor protein processing. Proc Natl Acad Sci U S A. 2005;102(17):6021–6026. doi:10.1073/pnas.0501823102
59. Inoue Y, Ando Y, Misumi Y, Ueda M. Current management and therapeutic strategies for cerebral amyloid angiopathy. Int J Mol Sci. 2021;22(8):3869. doi:10.3390/ijms22083869
60. Zellner A, Muller SA, Lindner B, et al. Proteomic profiling in cerebral amyloid angiopathy reveals an overlap with CADASIL highlighting accumulation of HTRA1 and its substrates. Acta Neuropathol Commun. 2022;10(1):6. doi:10.1186/s40478-021-01303-6
61. Fleckenstein M, Keenan TDL, Guymer RH, et al. Age-related macular degeneration. Nat Rev Dis Primers. 2021;7(1):31. doi:10.1038/s41572-021-00265-2
62. Deng Y, Qiao L, Du M, et al. Age-related macular degeneration: epidemiology, genetics, pathophysiology, diagnosis, and targeted therapy. Genes Dis. 2022;9(1):62–79. doi:10.1016/j.gendis.2021.02.009
63. Nashine S. Potential therapeutic candidates for Age-Related Macular Degeneration (AMD). Cells. 2021;10(9):2483. doi:10.3390/cells10092483
64. Dewan A, Liu M, Hartman S, et al. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science. 2006;314(5801):989–992. doi:10.1126/science.1133807
65. Deangelis MM, Ji F, Adams S, et al. Alleles in the HtrA serine peptidase 1 gene alter the risk of neovascular age-related macular degeneration. Ophthalmology. 2008;115(7):1209–15 e7. doi:10.1016/j.ophtha.2007.10.032
66. Jacobo SM, Deangelis MM, Kim IK, Kazlauskas A. Age-related macular degeneration-associated silent polymorphisms in HtrA1 impair its ability to antagonize insulin-like growth factor 1. Mol Cell Biol. 2013;33(10):1976–1990. doi:10.1128/MCB.01283-12
67. Iejima D, Nakayama M, Iwata T. HTRA1 overexpression induces the exudative form of age-related macular degeneration. J Stem Cells. 2015;10(3):193–203.
68. Gili P, Lloreda Martin L, Martin-Rodrigo JC, et al. Gene polymorphisms associated with an increased risk of exudative age-related macular degeneration in a Spanish population. Eur J Ophthalmol. 2022;32(1):651–657. doi:10.1177/11206721211002698
69. Vingerling JR, Dielemans I, Hofman A, et al. The prevalence of age-related maculopathy in the Rotterdam study. Ophthalmology. 1995;102(2):205–210. doi:10.1016/S0161-6420(95)31034-2
70. Chen W, Xu W, Tao Q, et al. Meta-analysis of the association of the HTRA1 polymorphisms with the risk of age-related macular degeneration. Exp Eye Res. 2009;89(3):292–300. doi:10.1016/j.exer.2008.10.017
71. Owen LA, Shirer K, Collazo SA, et al. The serine protease HTRA-1 is a biomarker for ROP and mediates retinal neovascularization. Front Mol Neurosci. 2020;13:605918. doi:10.3389/fnmol.2020.605918
72. Nakayama M, Iejima D, Akahori M, Kamei J, Goto A, Iwata T. Overexpression of HtrA1 and exposure to mainstream cigarette smoke leads to choroidal neovascularization and subretinal deposits in aged mice. Invest Ophthalmol Vis Sci. 2014;55(10):6514–6523. doi:10.1167/iovs.14-14453
73. Jones A, Kumar S, Zhang N, et al. Increased expression of multifunctional serine protease, HTRA1, in retinal pigment epithelium induces polypoidal choroidal vasculopathy in mice. Proc Natl Acad Sci U S A. 2011;108(35):14578–14583. doi:10.1073/pnas.1102853108
74. Iejima D, Itabashi T, Kawamura Y, et al. HTRA1 (high temperature requirement A serine peptidase 1) gene is transcriptionally regulated by insertion/deletion nucleotides located at the 3’ end of the ARMS2 (age-related maculopathy susceptibility 2) gene in patients with age-related macular degeneration. J Biol Chem. 2015;290(5):2784–2797. doi:10.1074/jbc.M114.593384
75. Tom I, Pham VC, Katschke KJ, et al. Development of a therapeutic anti-HtrA1 antibody and the identification of DKK3 as a pharmacodynamic biomarker in geographic atrophy. Proc Natl Acad Sci U S A. 2020;117(18):9952–9963. doi:10.1073/pnas.1917608117
76. Ahamed W, Yu RMC, Pan Y, et al. HTRA1 regulates subclinical inflammation and activates proangiogenic response in the retina and choroid. Int J Mol Sci. 2022;23(18):10206. doi:10.3390/ijms231810206
77. Tsuchiya A, Yano M, Tocharus J, et al. Expression of mouse HtrA1 serine protease in normal bone and cartilage and its upregulation in joint cartilage damaged by experimental arthritis. Bone. 2005;37(3):323–336. doi:10.1016/j.bone.2005.03.015
78. Kumar S, Nakashizuka H, Jones A, et al. Proteolytic degradation and inflammation play critical roles in polypoidal choroidal vasculopathy. Am J Pathol. 2017;187(12):2841–2857. doi:10.1016/j.ajpath.2017.08.025
79. Lu ZG, May A, Dinh B, et al. The interplay of oxidative stress and ARMS2-HTRA1 genetic risk in neovascular AMD. Vessel Plus. 2021;5. doi:10.20517/2574-1209.2020.48
80. Yanagisawa H, Schluterman MK, Brekken RA. Fibulin-5, an integrin-binding matricellular protein: its function in development and disease. J Cell Commun Signal. 2009;3(3–4):337–347. doi:10.1007/s12079-009-0065-3
81. Lin MK, Yang J, Hsu CW, et al. HTRA1, an age-related macular degeneration protease, processes extracellular matrix proteins EFEMP1 and TSP1. Aging Cell. 2018;17(4):e12710. doi:10.1111/acel.12710
82. Chen CY, Melo E, Jakob P, et al. N-Terminomics identifies HtrA1 cleavage of thrombospondin-1 with generation of a proangiogenic fragment in the polarized retinal pigment epithelial cell model of age-related macular degeneration. Matrix Biol. 2018;70:84–101. doi:10.1016/j.matbio.2018.03.013
83. Singla B, Aithbathula RV, Pervaiz N, et al. CD47 activation by thrombospondin-1 in lymphatic endothelial cells suppresses lymphangiogenesis and promotes atherosclerosis. Arterioscler Thromb Vasc Biol. 2023;43(7):1234–1250. doi:10.1161/ATVBAHA.122.318904
84. Meijles DN, Sahoo S, Al Ghouleh I, et al. The matricellular protein TSP1 promotes human and mouse endothelial cell senescence through CD47 and Nox1. Sci Signal. 2017;10(501). doi:10.1126/scisignal.aaj1784
85. Zhang L, Lim SL, Du H, et al. High temperature requirement factor A1 (HTRA1) gene regulates angiogenesis through transforming growth factor-beta family member growth differentiation factor 6. J Biol Chem. 2012;287(2):1520–1526. doi:10.1074/jbc.M111.275990
86. Asai-Coakwell M, French CR, Berry KM, et al. GDF6, a novel locus for a spectrum of ocular developmental anomalies. Am J Hum Genet. 2007;80(2):306–315. doi:10.1086/511280
87. Liu C, Xing F, He Y, et al. Elevated HTRA1 and HTRA4 in severe preeclampsia and their roles in trophoblast functions. Mol Med Rep. 2018;18(3):2937–2944. doi:10.3892/mmr.2018.9289
88. Friedrich U, Datta S, Schubert T, et al. Synonymous variants in HTRA1 implicated in AMD susceptibility impair its capacity to regulate TGF-beta signaling. Hum Mol Genet. 2015;24(22):6361–6373. doi:10.1093/hmg/ddv346
89. Yang Z, Camp NJ, Sun H, et al. A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science. 2006;314(5801):992–993. doi:10.1126/science.1133811
90. Saika S, Saika S, Liu CY, et al. TGFbeta2 in corneal morphogenesis during mouse embryonic development. Dev Biol. 2001;240(2):419–432. doi:10.1006/dbio.2001.0480
91. Maring JA, van Meeteren LA, Goumans MJ, Ten Dijke P. Interrogating TGF-β function and regulation in endothelial cells. Methods Mol Biol. 2016;1344:193–203.
92. Pan Y, Iejima D, Nakayama M, et al. Binding of Gtf2i-beta/delta transcription factors to the ARMS2 gene leads to increased circulating HTRA1 in AMD patients and in vitro. J Biol Chem. 2021;296:100456. doi:10.1016/j.jbc.2021.100456
93. Ng TK, Liang XY, Lai TY, et al. HTRA1 promoter variant differentiates polypoidal choroidal vasculopathy from exudative age-related macular degeneration. Sci Rep. 2016;6:28639. doi:10.1038/srep28639
94. Witmer A. Vascular endothelial growth factors and angiogenesis in eye disease. Prog Retinal Eye Res. 2003;22(1):1–29. doi:10.1016/S1350-9462(02)00043-5
95. Grisanti S, Tatar O. The role of vascular endothelial growth factor and other endogenous interplayers in age-related macular degeneration. Prog Retin Eye Res. 2008;27(4):372–390. doi:10.1016/j.preteyeres.2008.05.002
96. Martin DF, Maguire MG, Fine SL, et al.; Comparison of Age-related Macular Degeneration Treatments Trials Research G. Ranibizumab and bevacizumab for treatment of neovascular age-related macular degeneration: two-year results. Ophthalmology. 2012;119(7):1388–1398. doi:10.1016/j.ophtha.2012.03.053
97. Dedania VS, Grob S, Zhang K, Bakri SJ. Pharmacogenomics of response to anti-VEGF therapy in exudative age-related macular degeneration. Retina. 2015;35(3):381–391. doi:10.1097/IAE.0000000000000466
98. Wang Z, Zou M, Chen A, et al. Genetic associations of anti-vascular endothelial growth factor therapy response in age-related macular degeneration: a systematic review and meta-analysis. Acta Ophthalmol. 2021;100(3):e669–e680.
99. Lu Z, Lin V, May A, et al. HTRA1 synergizes with oxidized phospholipids in promoting inflammation and macrophage infiltration essential for ocular VEGF expression. PLoS One. 2019;14(5):e0216808. doi:10.1371/journal.pone.0216808
100. Wu L, Li X, Li Z, et al. HtrA serine proteases in cancers: a target of interest for cancer therapy. Biomed Pharmacother. 2021;139:111603. doi:10.1016/j.biopha.2021.111603
101. Xia J, Wang F, Wang L, Fan Q. Elevated serine protease HtrA1 inhibits cell proliferation, reduces invasion, and induces apoptosis in esophageal squamous cell carcinoma by blocking the nuclear factor-kappaB signaling pathway. Tumour Biol. 2013;34(1):317–328. doi:10.1007/s13277-012-0553-6
102. Baldi A, De Luca A, Morini M, et al. The HtrA1 serine protease is down-regulated during human melanoma progression and represses growth of metastatic melanoma cells. Oncogene. 2002;21(43):6684–6688. doi:10.1038/sj.onc.1205911
103. Chien J, Staub J, Hu SI, et al. A candidate tumor suppressor HtrA1 is downregulated in ovarian cancer. Oncogene. 2004;23(8):1636–1644. doi:10.1038/sj.onc.1207271
104. Chien J, Aletti G, Baldi A, et al. Serine protease HtrA1 modulates chemotherapy-induced cytotoxicity. J Clin Invest. 2006;116(7):1994–2004. doi:10.1172/JCI27698
105. He X, Ota T, Liu P, Su C, Chien J, Shridhar V. Downregulation of HtrA1 promotes resistance to anoikis and peritoneal dissemination of ovarian cancer cells. Cancer Res. 2010;70(8):3109–3118. doi:10.1158/0008-5472.CAN-09-3557
106. Halberg N, Sengelaub CA, Navrazhina K, Molina H, Uryu K, Tavazoie SF. PITPNC1 recruits RAB1B to the golgi network to drive malignant secretion. Cancer Cell. 2016;29(3):339–353. doi:10.1016/j.ccell.2016.02.013
107. Ding X, Ji J, Jiang J, et al. HGF-mediated crosstalk between cancer-associated fibroblasts and MET-unamplified gastric cancer cells activates coordinated tumorigenesis and metastasis. Cell Death Dis. 2018;9(9):867. doi:10.1038/s41419-018-0922-1
108. Benedito R, Roca C, Sorensen I, et al. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137(6):1124–1135. doi:10.1016/j.cell.2009.03.025
109. Lu H, Yuan P, Ma X, et al. Angiotensin-converting enzyme inhibitor promotes angiogenesis through Sp1/Sp3-mediated inhibition of notch signaling in male mice. Nat Commun. 2023;14(1):731. doi:10.1038/s41467-023-36409-z
110. Fasoulakis Z, Galazios G, Koukourakis M, et al. Expression of Delta Like Ligand 4 (DLL4) in endometrial carcinomas and tumor vasculature. J BUON. 2021;26(4):1327–1332.
111. Wu F, Yang J, Liu J, et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct Target Ther. 2021;6(1):218. doi:10.1038/s41392-021-00641-0
112. Chien J, Ota T, Aletti G, et al. Serine protease HtrA1 associates with microtubules and inhibits cell migration. Mol Cell Biol. 2009;29(15):4177–4187. doi:10.1128/MCB.00035-09
113. Tang T, Zhu Q, Li X, et al. Protease Nexin I is a feedback regulator of EGF/PKC/MAPK/EGR1 signaling in breast cancer cells metastasis and stemness. Cell Death Dis. 2019;10(9):649. doi:10.1038/s41419-019-1882-9
114. Baker AM, Bird D, Welti JC, et al. Lysyl oxidase plays a critical role in endothelial cell stimulation to drive tumor angiogenesis. Cancer Res. 2013;73(2):583–594. doi:10.1158/0008-5472.CAN-12-2447
115. Wang N, Eckert KA, Zomorrodi AR, et al. Down-regulation of HtrA1 activates the epithelial-mesenchymal transition and ATM DNA damage response pathways. PLoS One. 2012;7(6):e39446. doi:10.1371/journal.pone.0039446
116. Colak S, Ten Dijke P. Targeting TGF-β signaling in cancer. Trends Cancer. 2017;3(1):56–71. doi:10.1016/j.trecan.2016.11.008
117. Tang H, Leung L, Saturno G, et al. Lysyl oxidase drives tumour progression by trapping EGF receptors at the cell surface. Nat Commun. 2017;8(1):14909. doi:10.1038/ncomms14909
118. Larson C, Oronsky B, Carter CA, et al. TGF-beta: a master immune regulator. Expert Opin Ther Targets. 2020;24(5):427–438. doi:10.1080/14728222.2020.1744568
119. Shi X, Yang J, Deng S, et al. TGF-β signaling in the tumor metabolic microenvironment and targeted therapies. J Hematol Oncol. 2022;15(1):135. doi:10.1186/s13045-022-01349-6
120. Tsai FC, Chen YL, Yen KC, et al. Gene expression changes of humans with primary mitral regurgitation and reduced left ventricular ejection fraction. Int J Mol Sci. 2021;22(7):3454. doi:10.3390/ijms22073454
121. Muratoglu SC, Belgrave S, Hampton B, et al. LRP1 protects the vasculature by regulating levels of connective tissue growth factor and HtrA1. Arterioscler Thromb Vasc Biol. 2013;33(9):2137–2146. doi:10.1161/ATVBAHA.113.301893
122. Quinn C, Rico MC, Merali C, et al. Secreted folate receptor γ drives fibrogenesis in metabolic dysfunction-associated steatohepatitis by amplifying TGFβ signaling in hepatic stellate cells. Sci Transl Med. 2023;15(715):eade2966. doi:10.1126/scitranslmed.ade2966
123. Sabino F, Egli FE, Savickas S, et al. Comparative degradomics of porcine and human wound exudates unravels biomarker candidates for assessment of wound healing progression in trauma patients. J Invest Dermatol. 2018;138(2):413–422. doi:10.1016/j.jid.2017.08.032
124. Jin SF, Wang YX, Xu N, et al. High temperature requirement factor A1 (HTRA1) regulates the activation of latent TGF-β1 in keloid fibroblasts. Cell Mol Biol. 2018;64(1):107–110. doi:10.14715/cmb/2018.64.2.19
125. Lorenzi M, Lorenzi T, Marzetti E, et al. Association of frailty with the serine protease HtrA1 in older adults. Exp Gerontol. 2016;81:8–12. doi:10.1016/j.exger.2016.03.019
126. Xu W, Liu X, Han W, et al. Inhibiting HIF-1 signaling alleviates HTRA1-induced RPE senescence in retinal degeneration. Cell Commun Signal. 2023;21(1):134. doi:10.1186/s12964-023-01138-9
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.