")
Back to Journals » International Journal of Nanomedicine » Volume 19
Regulating Cholesterol in Tumorigenesis: A Novel Paradigm for Tumor Nanotherapeutics
Authors Wu H, Wu X, Zhao M , Yan J, Li C, Zhang Z, Tang S, Wang R, Fei W
Received 12 September 2023
Accepted for publication 23 January 2024
Published 1 February 2024 Volume 2024:19 Pages 1055—1076
DOI https://doi.org/10.2147/IJN.S439828
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Jie Huang
Huifeng Wu,1 Xiaodong Wu,2 Mengdan Zhao,3 Jingjing Yan,3 Chaoqun Li,3 Zhewei Zhang,1 Sangsang Tang,2 Rong Wang,3 Weidong Fei3
1Department of Urology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, People’s Republic of China; 2Department of Gynecologic Oncology, Women’s Hospital, Zhejiang University School of Medicine, Hangzhou, 310006, People’s Republic of China; 3Department of Pharmacy, Women’s Hospital, Zhejiang University School of Medicine, Hangzhou, 310006, People’s Republic of China
Correspondence: Weidong Fei; Rong Wang, Email [email protected]; [email protected]
Abstract: During the past decade, “membrane lipid therapy”, which involves the regulation of the structure and function of tumor cell plasma membranes, has emerged as a new strategy for cancer treatment. Cholesterol is an important component of the tumor plasma membrane and serves an essential role in tumor initiation and progression. This review elucidates the role of cholesterol in tumorigenesis (including tumor cell proliferation, invasion/metastasis, drug resistance, and immunosuppressive microenvironment) and elaborates on the potential therapeutic targets for tumor treatment by regulating cholesterol. More meaningfully, this review provides an overview of cholesterol-integrated membrane lipid nanotherapeutics for cancer therapy through cholesterol regulation. These strategies include cholesterol biosynthesis interference, cholesterol uptake disruption, cholesterol metabolism regulation, cholesterol depletion, and cholesterol-based combination treatments. In summary, this review demonstrates the tumor nanotherapeutics based on cholesterol regulation, which will provide a reference for the further development of “membrane lipid therapy” for tumors.
Keywords: cholesterol, tumor, plasma membrane, nanotherapeutics
Graphical Abstract:
Introduction
Cholesterol plays a vital role in regulating mammalian survival and development. In addition to being a bile acid and steroid hormone precursor, cholesterol acts as a fundamental component of the plasma membrane, serving an essential function in maintaining cellular structure and biological activities.1 Cholesterol metabolism is often dysregulated in cancer cells. Owing to their accelerated cell proliferation, cancer cells have a higher demand for cholesterol, which is used in membrane synthesis and various other cellular processes. Tumor cells exhibit marked alterations in cholesterol homeostasis.2 This includes upregulation in cholesterol synthetic genes, increased cholesterol uptake via low-density lipoprotein receptors, and abnormal cholesterol metabolism. These alterations lead to elevated cellular cholesterol levels, which in turn contribute to the initiation and progression of cancer.2–5 Moreover, cholesterol plays an important role in inhibiting immune-effector cells and antigen presentation. Both preclinical and clinical studies have demonstrated that altering cholesterol metabolism strongly suppresses tumor development, reconfigures immunological profile, and reinforces antitumor immunity.6
In clinical practice, the upregulation of cholesterol levels in patients with cancer is a common observation,2 and increased circulating cholesterol is directly correlated with the development of some cancers.7–9 For example, an increase of 10 mg/dL in cholesterol is related to a 9% higher risk of prostate cancer recurrence.9 In sarcoma, acute myeloid leukemia and melanoma, augmented cholesterol synthetic activity has been associated with reduced overall survival rate of patients. However, in low-grade gliomas, it appears to be associated with prolonged patient survival.10 The Cancer Genome Atlas indicated that upregulated cholesterol biosynthetic genes are strongly associated with shorter overall survival among patients with tumors such as ovarian cancer.10 In general, abnormal cholesterol metabolism greatly promotes the occurrence and development of cancers, influencing processes like cell proliferation, migration, and invasion.6,11,12 Therefore, reducing cholesterol synthesis or blocking cholesterol transport can inhibit the growth and invasion of tumor cells.13–15
Over the past 10 years, many researchers have conducted studies on antitumor therapies by depleting cholesterol, and a series of effective small molecule drugs against tumors have been identified in preclinical and clinical studies (Table 1). The therapeutic mechanisms of these small molecules mainly include cholesterol synthesis interference (eg, statins, bisphosphonates, and terbinafine), inhibition of cholesterol transport (eg, ezetimibe, GRX-104, and itraconazole), and regulation of cholesterol metabolism (eg, ABTL0812). It is worth noting that, unlike other specific molecules in tumor sites, cholesterol is a critical plasma membrane constituent in all cells. Therefore, targeted regulation of cholesterol at the tumor site is remarkable in reducing the impact on normal cells.
 |
Table 1 Clinical Trials of Various Cholesterol-Lowering Drugs as a Tumor Therapeutic Strategy |
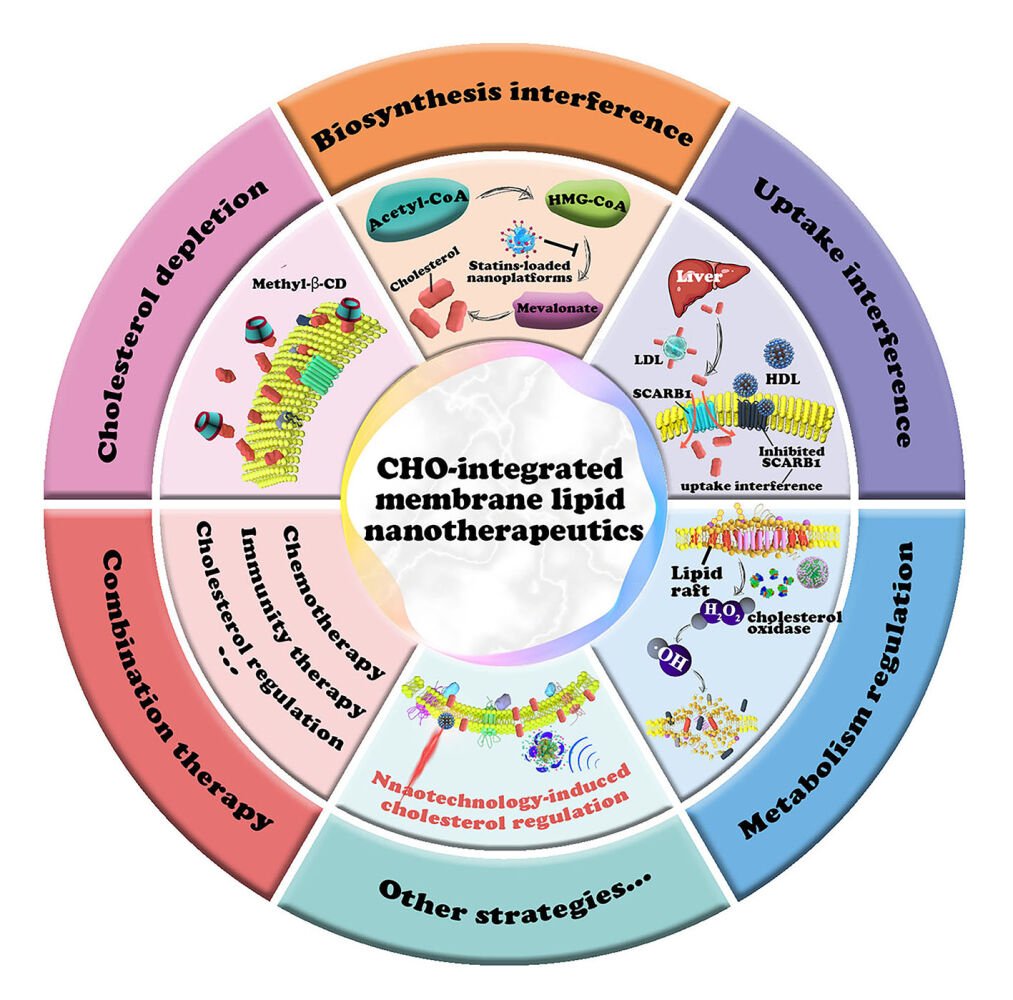
Rapid progress in nanoscience creates novel opportunities for tumor treatment.16–18 The nanocarriers can accumulate in tumor tissues through the enhanced permeability and retention (EPR) effect and/or actively targeting effect via surface modifications.19 Nanocarriers containing cholesterol-regulating drugs mitigate the adverse effects of drugs on blood cells during the circulation. Furthermore, nanoplatforms interact with the plasma membrane before undergoing cellular endocytosis, providing them with an inherent advantage in the field of membrane lipid therapy focused on cholesterol regulation.20 For example, liposomes always release drugs around the plasma membrane by merging with the membrane,21 and the released molecules could regulate the structure and function of the plasma membrane. In recent years, some studies have reviewed the association between abnormal cholesterol metabolism and tumor development, while summarizing the tumor treatment strategies by targeting cholesterol.6,22 However, there are few comprehensive reviews that have summarized the significance of nanotechnology in targeted interventions for membrane lipid modulation based on cholesterol regulation. To demonstrate the effectiveness of nanotherapeutics based on cholesterol regulation and highlight the importance of such research in this area, this review introduces cholesterol homeostasis within the body and demonstrates the association between overexpressed cholesterol with proliferation, metastasis, drug resistance of tumor cells, and influence on tumor immune microenvironment. This section will provide a theoretical basis for finding highly effective molecular targets and designing therapeutic approaches for cholesterol-integrated membrane lipid therapy. The paper further focuses on the current nanotherapeutic strategies based on cholesterol regulation and demonstrates the advantages of nanotechnology in cholesterol-related tumor membrane lipid therapy (Figure 1). Moreover, we discuss the potential clinical applications of cholesterol interference strategies in cancer therapy, an area that is rarely presented in other relevant reviews. In summary, this review illustrates the association between the overexpressed cholesterol and tumorigenesis, while also highlighting the advantages of nanotechnology in the regulation of cholesterol in plasma membrane.
 |
Figure 1 An overview of plasma membrane cholesterol regulation tumor nanotherapeutics. Abbreviations: Acetyl-CoA, Acetoacetyl coenzyme A; HMG-CoA, 3-hydroxy-3-methyl glutaryl coenzyme A; LDL, low-density lipoprotein; HDL, high-density lipoprotein; SCARB1, scavenger receptor type B1. |
Cholesterol-Related Tumorigenesis and Progression
Cholesterol Homeostasis
Cholesterol in mammals primarily comes from two sources including endogenous synthesis in the liver and dietary absorption.2 It is transported through the bloodstream to cells throughout the body in the form of low-density lipoprotein (LDL) knots.10 The free cholesterol within cells moves across the plasma membrane and membrane-associated organelles.1,10 Cholesterol in the body mainly exists as free cholesterol and cholesteryl ester, both of which are widely distributed in various tissues of the body. Among them, the brain, nerve tissues, and glands of steroid hormones (such as adrenal glands and gonads) have higher cholesterol content, whereas muscle tissues generally contain lower levels of cholesterol.
Cholesterol homeostasis is closely controlled by an intricate network of proteins.1 Among them, sterol regulatory element binding protein transcription factor 2 and liver X receptor (LXR) are key cholesterol homeostatic modulators.1 Endoplasmic reticulum-associated cholesterol functions as a sensor for intracellular cholesterol homeostasis. The reduction of endoplasmic reticulum cholesterol induces the translocation of sterol regulatory element binding protein transcription factor 2 from endoplasmic reticulum to Golgi, which in turn translocates to the nucleus to transcribe cholesterol synthesis- and uptake-related genes (eg, hydroxy-3-methylglutaryl-CoA reductase (HMGCR) and LDL receptors).1 Alternatively, enhanced intracellular cholesterol content inhibits cholesterol production while facilitating its efflux through the activation of the LXR by oxidized sterols, an oxidized derivative of cholesterol.23 Owing to their accelerated cell proliferation, cancer cells always have a great demand for cholesterol, which is used in membrane synthesis and other vital cellular functions.6 The overexpressed cholesterol can modulate tumorigenic and cancer progression-related networks via covalent protein modification and facilitation of specialized membrane microdomain production.22,24 The following section focuses on the role of cholesterol in key events of tumor development to explore more therapeutic targets and treatment strategies in greater detail (Figure 2).
 |
Figure 2 Schematic diagram of cholesterol-related tumorigenesis and progression. (A) The role of cholesterol in tumor cell proliferation. (B) Cholesterol-mediated regulation of tumor invasion and metastasis. (C) Cholesterol-mediated regulation of tumor drug resistance. (D) The mechanisms of cholesterol in suppressing the tumor immune microenvironment. Abbreviations: GPCRs, G-protein-coupled receptors; mTORC1, mammalian target of rapamycin C1; STARD3, steroidogenic acute regulatory related lipid transfer domain-3; SREBP2, sterol regulatory element-binding protein 2; TAM, tumor-associated macrophages; 27-HC, 27-hydroxycholesterol. |
The Role of Cholesterol in Tumor Cell Proliferation
Cholesterol promote the proliferation of tumor cells through the interaction with receptors on the plasma membrane (Figure 2A). For instance, when cholesterol binds to specific sites on G-protein-coupled receptors (GPCR) like the adenosine A2A receptor, or GPCR-like transducers such as the Smoothened (Smo) receptors, it can promote the proliferation of tumor cells.25 Numerous clinical trials have been conducted to test the efficacy of Smo inhibitors in the treatment of tumors (such as NCT05177770 and NCT01218477). Findings from these trials have shown that Smo inhibitors could effectively delay the progression of tumors. It has been reported that Smo inhibitors can competitively inhibit the binding of cholesterol to Smo receptors, thereby inhibiting signaling pathways that enhance tumor growth. Table 1 outlines some compounds (resveratrol and GRX-104) that modulate the proliferation of tumor cells by regulating cholesterol content. For instance, resveratrol suppress the synthesis of cholesterol by blocking sterol regulatory element-binding protein 2 (SREBP2), thereby reducing cholesterol levels and delaying the proliferation of tumor cells.26 Another membrane receptor, LXR, regulates the metabolism of cholesterol by transporting cholesterol out of the cell. Studies have suggested that overactivation of LXR could enhance cholesterol efflux, which precipitated anti-proliferative effects and inhibited tumor proliferation.27,28
Lipid raft (LR), which contains plentiful cholesterol and sphingolipids, is an important structural domain of the plasma membrane.29 LR is located on cell membranes and facilitates carcinogenic cellular networks.30 Dysregulated cholesterol content can potentially alter LR configuration, leading to the change of essential molecules such as receptors, calcium channels, and protein kinases.31 Among the associated protein kinases, Akt is strongly correlated with LR. Once activated, it relocates to LR whereby it enhances the proliferation of tumor cells.31,32 Similarly, the methyl-β-cyclodextrin (MβCD, a cholesterol chelator)-based LR damage strongly induces cellular apoptosis by activating the transcription of common apoptotic bioindicators.33 Generally, cholesterol can recognize receptors and activate the downstream signaling cascade for tumor proliferation.
Among various cancers, prostate cancer stands out as having the most evident association with overexpressed cholesterol, which fosters its occurrence and progression.9 Fatty acid and cholesterol production is markedly enhanced in prostate cancer cells. In mice bearing prostate cancer cells, the rise of serum cholesterol could enhance the cholesterol levels in the LR of tumor cells, change the LR-mediated downstream signal conduction, and promote the proliferation and growth of tumors.34 Long-term statin medication has been associated with a lower risk of prostate cancer.35 Cholesterol can also be negatively regulated in cancer cells by reducing cholesterol absorption. For instance, the FDA-approved drug ezetimibe suppresses the proliferation of prostate tumor cells via inhibition of intestinal cholesterol absorption.2
Cholesterol-Mediated Regulation of Tumor Invasion and Metastasis
In addition to its role in tumor cell proliferation, cholesterol also serves critical functions in tumor invasion and metastasis (Figure 2B). Emerging evidence suggested that lysosomal cholesterol stimulated the mammalian target of rapamycin C1 (mTORC1),36,37 which in turn promoted the proliferation, infiltration, and metastasis of tumor cells.37 Moreover, mitochondrial inner membrane-related cholesterol also possesses physiological activities, such as bile acid and steroid hormone biosynthesis.37 Multiple reports have suggested that cholesterol import was strictly regulated by two critical proteins in mitochondria, namely, steroidogenic acute regulatory (STAR) and STAR-related lipid transfer domain-3 (STARD3).38,39 Elevated STARD3 levels augment cell proliferation, while an excess of STARD3 reduces tumor adhesiveness, therefore enabling tumor invasion and metastasis.10,39
Cell adhesion following tumor cell metastasis is the most important step in ectopic engraftment.40 Cholesterol-rich LR possesses abundant cancer-associated axes and cell adhesion molecules, which in turn promote the metastasis of cancer cells. For instance, CD44, a cell adhesion protein located in LR, is overexpressed in tumor cells and modulates tumor infiltration and metastasis.41 LRs rich in cholesterol are rigid and support CD44 located within them, endowing CD44 physiological activity.42 MβCD could induce the shedding of CD44 in tumor cells and inhibit the invasion and metastasis of tumor cells.2
Cholesterol metabolites, such as 27-hydroxycholesterol (27-HC), play an important role in cancer development. In breast cancer, 27-HC acts as an estrogen receptor agonist, promoting tumor growth and metastasis.43 Statins have antitumor effects, particularly against mesenchymal cancer cells. When combined with chemotherapy drugs, statins could effectively inhibit the activity of tumor cells that have undergone epithelial-mesenchymal transition, and thus suppress the metastasis of tumor cells.44 In many clinical studies, statins have been used to inhibit the progression of breast cancer, colorectal cancer, and prostate cancer by suppressing the metastasis and invasion of tumors (Table 1).
Cholesterol-Mediated Regulation of Tumor Drug Resistance
In adverse external environments, cancer cells always activate cholesterol production, which in turn benefits their adaptation and survival. Figure 2C depicts the reported mechanisms of cholesterol-mediated tumor drug resistance. Drug-resistant cell plasma membranes display higher levels of cholesterol compared to drug-sensitive cells.45–47 The abundant plasma membrane-associated cholesterol in drug-resistant cancer cells leads to a more rigid plasma membrane, resulting in reduced drug permeability. This is one of the key factors contributing to the development of tumor drug resistance.45,48,49 LRs are specialized domains within the plasma membrane enriched in cholesterol and sphingomyelin. They play an important role in various cellular activities such as endocytosis and nutrient transport.50 The drug-resistant phenotype of tumors is highly correlated with the upregulation of LR contents.24 Increased cholesterol and sphingomyelin levels in the LR of drug-resistant cancer cells enhance the content, recycling, and bioactivity of multidrug resistance transporters (ABC transporters) localized in these regions,51,52 thus accelerating the extracellular transport of chemotherapeutic drugs. Further, cholesterol can change the stiffness and fluidity of LR, thereby changing the spatial conformation of the drug-resistant protein in the structural domain. This facilitates the binding of drug-resistant proteins to intracellular chemotherapeutic drugs, allowing for their transportation out of the cells.53 Maintaining the high levels of cholesterol in LR of drug-resistant cells is essential to facilitate the biological activity of associated p-glycoprotein (p-gp).54 Depletion of cholesterol-sphingomyelin-rich LR with a small molecule drug (5-aza-2’-deoxycytidine) could successfully reverse drug resistance in tumors.55
In addition to the above two aspects, isoprene produced in the cholesterol production process can act as a second messenger of cell signalings. It triggers cascading signals related to multiple drug resistance, stimulates the transcription of drug efflux transporter genes, and enhances metabolic reprogramming in drug-resistant cells.56 For example, higher cholesterol levels in drug-resistant ovarian cancer cells can simultaneously upregulate the expression of p-gp and liver X receptor β, leading to resistance against cisplatin-mediated apoptosis. However, cholesterol clearance through MβCD or silencing of liver X receptor β could restore the sensitivity of drug-resistant ovarian cells to chemotherapeutic drugs.57,58 Moreover, isoprenoids produced in the mevalonate pathway can be integrated into the ubiquinone hydrophobic tail to increase the biosynthesis of adenosine triphosphate (ATP) to provide energy for ABC transporters.59 In previous studies, it was found that statins inhibited cholesterol synthesis and reversed resistance to paclitaxel in lung cancer. In addition, the cholesterol scavenger (acetyl plumbagin) reversed the resistance of breast cancer to tamoxifen.60,61 MβCD-mediated cholesterol clearance strategy has been proposed to reverse drug resistance by damaging the structure of the lipid raft.51 Such strategies have shown promising results in controlling tumor resistance in clinical practice. In general, abnormal cholesterol metabolism in plasma membrane promotes the development of drug resistance in cancers by limiting the entry of drugs into tumor cells, regulating the expression/functions of drug resistance proteins, and activating multiple drug resistance cascade signals. Based on the above analysis, depleting plasma membrane cholesterol is a possible approach for drug-resistant cancer therapy.20
The Role of Cholesterol in Suppressing the Tumor Immune Microenvironment
Recent studies demonstrated that cholesterol served a crucial function in remodeling tumor immune microenvironment (Figure 2D).62 Specifically, the membranal cholesterol level influences T-cell activity and function,63 and downstream cholesterol products are essential for B-cell migration.64 A study reported in “Nature” suggested that inhibiting PCSK9, a protein that regulates cholesterol metabolism, could promote tumor response to immune-check-point therapy by enhancing major histocompatibility complex class I protein expression on the surface of tumor cells. Such effect upregulated the invasion of intratumoral cytotoxic T cells.65 Thus, regulating cholesterol metabolism is an effective strategy for tumor immunotherapy.
Abnormal cholesterol metabolism impairs the effectiveness of CD8+ T cells.66 Genes related to cholesterol synthesis and transport are significantly upregulated in early-stage breast cancer or melanoma cocultures. Moreover, augmented cholesterol ester levels greatly stimulate the senescence of effector CD8+ T cells.67 Similarly, a study published in “Cell Metabolism” suggested that the uptake of free cholesterol by CD8+ T cells induced the stress of the endoplasmic reticulum, which eventually led to the exhaustion of CD8+ T cells, thus reducing tumor responsiveness to immune checkpoint inhibitors (ICIs) significantly.68 In 2020, a study published in “Nature Metabolism” demonstrated that free cholesterol in tumor immune microenvironment recruited suppressive immune cells, thereby inducing strong antitumor immunosuppression and accelerating tumor cell immune escape.6 A high cholesterol content inhibits the activation of SREBP2 and SREBP2 drives the mevalonate pathway. Subsequently, the decreased intermediates such as farnesyl pyrophosphate and geranylgeranyl pyrophosphate serve as substrates for prenylation, affecting interactions with intracellular pathways that play an important role in maintaining the biological activity of T cells.69
Tumor-infiltrating immune cells, such as neutrophils, tumor-associated macrophages (TAMs), and dendritic cells undergo reprogramming due to alterations in cholesterol metabolism. In the presence of a high-cholesterol diet, 27-HC recruits polymorphonuclear neutrophils and γδ T cells to downregulate cytotoxic CD8+ T cells, thereby augmenting tumor metastasis in a breast cancer model.70 Cancer cell-mediated hyaluronic acid oligomer secretion increases cholesterol efflux from TAMs, directing them towards an M2-like phenotype which further promotes tumor progression.71 Improving the response rate of ICIs is an urgent challenge in the field of tumor immunotherapy. Over the past five years, cholesterol and associated metabolites have attracted greater scientific significance for reshaping the tumor immune microenvironment. Clinically, retrospective studies can be employed for investigating the impact of cholesterol-lowering drugs on the response rate and efficacy of ICIs. If the response rate of ICIs can be improved by cholesterol-lowering drugs, it will potentially mark a new era of tumor immunotherapy.
The above discussion provides a theoretical basis for the formulation of new clinical treatment strategies. Firstly, considering that cholesterol is strongly related to tumor metastasis, drug resistance, and influences the immunosuppressive microenvironment, tumor treatment bottlenecks encountered in clinical practice could be overcome from the perspective of regulating cholesterol metabolism. Secondly, the majority of clinical trials have mainly tested drugs that inhibit the synthesis of cholesterol. This paper demonstrated that the prevention of cholesterol uptake and promotion of cholesterol efflux/consumption can also suppress the tumorigenesis effectively. The discussion in this section suggests that drugs that target various aspects of cholesterol metabolism are potential agents in cancer therapy.
Cholesterol-Integrated Tumor Nanotherapeutics
As discussed in Cholesterol-Related Tumorigenesis and Progression, the alteration in cholesterol level of tumor cells is a potential strategy for tumor therapy. Compared with other antitumor drugs, cholesterol regulating agent requires more potential tumor targeting effect, as cholesterol is also one of the major components of normal tissue cell membranes. Results shown in Table 2 suggest that cholesterol-regulating nanoplatforms can be functionalized with tumor-targeting ligands (such as CD44, RGD, folic acid, and transferrin) to achieve targeted therapy.72,73 The particle size of nanoplatforms designed to regulate cholesterol levels predominantly ranged from 1 to 100 nm. Nanopreparations featuring smaller particle sizes exhibit enhanced cellular uptake and improved deep tumor tissue penetration. Table 2 outlines a variety of nanomaterials, comprising inorganic and organic substances. Notably, some organic materials possess inherent cholesterol-regulating capabilities. For instance, artificial high-density lipoprotein nanoparticles (HDL NPs) can bind to scavenger receptor type B1 (SCARB1) and suppress the uptake of cholesterol, while MβCD can absorb and deplete cholesterol in tumor cells. In the following section, the existing nanotechnology-based cholesterol regulation strategies (Table 2) and their therapeutic effects on tumors will be introduced in detail.
 |
Table 2 The Classification and Mechanisms of Cholesterol-Depleting Nanoplatforms in Cancer Therapy |
Interference of Cholesterol Biosynthesis
Inhibiting Cholesterol Biosynthesis by Statins
Cholesterol is synthesized through the mevalonate pathway, which consists of three stages: acetyl CoA synthesis of isopentane pyrophosphate, squalene synthesis, and squalene conversion to cholesterol. HMGCR and squalene epoxidase are major cholesterol biosynthetic enzymes.6,91 Blocking cholesterol biosynthesis is a promising and highly effective strategy for cancer intervention. Statins, as competitive HMGCR inhibitors, are commonly used as lipid-depleting drugs. They effectively suppress the activity of HMGCR, thus blocking the mevalonate pathway. The use of statins has been intricately linked to reduced risk of melanoma, non-Hodgkin’s lymphoma, endometrial cancer, and breast cancer.10 A randomized controlled clinical trial (NCT04705909) investigated the effect of pitavastatin combined with neoadjuvant chemotherapy on breast cancer patients. In this study, 70 female patients with pathologically-proven invasive breast cancer were randomized to receive with or without pitavastatin once daily concomitantly with standard neoadjuvant chemotherapy protocols for 6 months. It was observed that patients in the pitavastatin group had significantly higher median reduction in tumor size compared with those in the control group. Subgroup analysis of the pitavastatin group revealed that patients with positive human epidermal growth receptor 2 had higher median reduction in Ki67 analysis relative to those with negative human epidermal growth receptor 2.92 These results demonstrated that regulating cholesterol biosynthesis may be an effective strategy for treating breast cancer patients. Table 1 displays several clinical programs (NCT02026583, NCT04705909, NCT02569645, NCT00416403 and NCT01555632) performed to test the efficacy of statins in different tumors.
After reviewing the results of the above clinical studies, our groups found that statins alone have failed to demonstrate satisfactory tumor suppressive effects. Many studies also have explored the tumor therapeutic effect of statins combined with other anticancer drugs. In one research, researchers designed a liposome for the co-delivery of simvastatin (SV) and paclitaxel (PTX) for combating drug-resistant lung cancer (Figure 3A).61 The liposome modified with cell-penetrating peptide delivered SV to block cholesterol biosynthesis of tumor cells. Results of the study demonstrated that drug resistance of A549T cells was reversed by SV (Figure 3B). It can be seen in Figure 3C that A549T cells formed stronger rapid fibronectin adhesion to fibronectin, an integrin β3-binding extracellular matrix, as opposed to parental A549 cells. The above results indicated that SV abrogated the integrin β3 and fibronectin association in A549T cells, therefore re-sensitizing cells to PTX. When treated with SV, the expression of integrin β3 and the level of LR in tumor cells significantly decreased (Figure 3D and E). Results demonstrated that SV repolarized M2 to M1 phenotype, by downregulating the M2 biomarker CD206 and a protumor cytokine TGF-β (Figure 3F and G). In animal models, multidrug-loaded liposomes were found to have the best antitumor effect (Figure 3H). It suggested that SV regulated macrophage polarization and epithelial-mesenchymal transition (EMT) in cancer cells by blocking cholesterol synthesis. As demonstrated in Cholesterol-Mediated Regulation of Tumor Drug Resistance, cholesterol is essential for maintaining the function of p-gp multidrug efflux pumps.93 In conclusion, regulating cholesterol metabolism could reverse EMT and repolarize TAM to treat drug-resistant cancer, and the new application of statins has significant clinical value against tumor drug resistance. In another study, Guo et al designed and developed a low density lipoprotein receptor related protein 1-upregulated nanoparticles system for the codelivery of SV and doxorubicin (DOX) to multifocal and infiltrative brain cancer.79 The results demonstrated that the inhibition of HMGCR led to the consumption of intracellular cholesterol and the activity decrease of p-gp for brain cancer therapy.
 |
Figure 3 (A) Schematic illustrating the effect of multifunctional liposome on drug-resistance tumor. (B) SV sensitized A549T cells to PTX. (C) Adherent fractions of fibronectin (FN) and cationic poly-L-lysine (PLL) in A549T and A549 cells. (D and E) Integrin β3 level and lipid raft formation in A549T cells were reduced by SV. (F) Quantitative analysis of CD206 expression. (G) SV suppressed CD206 and TGF-β1 expression in M2. (H) Tumor growth of A549T tumors after different treatments. *p < 0.05, **p < 0.01, ***p < 0.001, and n.s. = no significant difference. Abbreviations: TNF-α, an M1-associated marker; CD206, an M2 marker; SV, simvastatin; PTX, paclitaxel. Notes: Reproduced from Jin, H, He Y, Zhao P, et al Targeting lipid metabolism to overcome EMT-associated drug resistance via integrin beta3/FAK pathway and tumor-associated macrophage repolarization using legumain-activatable delivery. Theranostics. 2019; 9(1): 265–278. Creative Commons.61 |
Inhibiting Cholesterol Biosynthesis by Other Pathways
Cholesterol synthesis is regulated by a complex interplay of transcriptional and post-translational regulatory systems. Among them, SREBP2 is the principal transcriptional regulator responsible for governing cholesterol production.91 SREBP2 is regarded as a sensor that regulates lipid homeostasis. When cholesterol levels are low, an imbalance in SREBP2 appears and the SREBP2 pathway is activated to synthesize cholesterol. In clinical trials (NCT00127205, NCT03295981 and NCT02366884), bisphosphonates and terbinafine inhibited cholesterol synthesis by inhibiting farnesyl diphosphate synthase (FDPS) or squalene epoxidase. Dai et al employed a phototherapy approach mediated by black TiO2 nanoprobe to reduce intracellular lipid levels by modulating cholesterol pathways, as part of their strategy for treating atherosclerosis.80 In another study, Rink et al designed a functional lipoprotein nanoparticle for the specific targeting of cellular cholesterol in diffuse large B-cell lymphoma (DLBCL).76 In their study, HDL nanoparticles combined with ibrutinib showed remarkable therapeutic effects in SUDHL4 and TMD8 cells by inhibiting cholesterol synthesis and increasing cholesterol depletion. Functional nanoparticles can target the delivery of drugs to tumors and reduce the side effects. Therefore, incorporating ibrutinib into functional nanoparticles may offer an improved approach to inhibiting cholesterol synthesis in cancer cells. The content of this section inspired us that nanotechnology could make cholesterol blocking more efficient, and can realize the combined treatment of cholesterol synthesis inhibitors and cytotoxic drugs.
Cholesterol Uptake Interference
After synthesized in the liver, cholesterol is transported to the outside of the liver by binding to high-density lipoprotein. In clinical trials (NCT02534376 and NCT02749513), ezetimibe and itraconazole could block cholesterol uptake by targeting Niemann-Pick C1 protein (NPC1) for tumor therapy. In another clinical trial (NCT02922764), GRX-104 increased cholesterol export by targeting LXR for endometrial cancer and lung cancer therapy. During the past two decades, there has been a growing interest among researchers in designing HDL-mimicking nanotherapeutics. These nanotherapeutics are intended to serve as a supplemental therapeutic intervention for cardiovascular disease patients who are already consuming drugs to lower atherogenic lipoproteins.94 Similar to cardiovascular disease, the progression of adrenocortical carcinoma is closely related to cholesterol. The bio-inspired HDL nanoparticles, which specifically bind to the SCARB1, can disrupt cellular cholesterol and cholesteryl ester homeostasis by inhibiting free cholesterol uptake through the SCARB1,74,95 which results in the depletion of cellular cholesterol and cholesteryl esters.95,96 For cells rich in cholesterol such as SUDHL4 cells, a reduction in cellular cholesterol could cause apoptosis after incubation with HDL nanoparticles in a dose-dependent manner.78 Thus, SCARB1 is a potential target for dysregulated cholesterol homeostasis.95–98
In a study, Rink et al employed HDL-NPs for DLBCL treatment by directly blocking SCARB1-mediated cholesterol uptake (Figure 4A).76 The results showed that all B-cell lymphoma cell lines exhibited dose-dependent cellular death upon exposure to HDL-NPs (Figure 4B). It was further shown that germinal center DLBCL line SUDHL4 was more sensitive to HDL-NPs than activated B cell DLBCL lines (Figure 4B). They further investigated the HDL-NPs-mediated regulation of cholesterol efflux from DLBCL lines (Figure 4C and D). It was found that following HDL NPs exposure, both SUDHL4 and TMD8 cells released cholesterol. The blocking antibodies partially disrupted the association of HDL-NPs with cholesterol efflux, a phenomenon that was not observed in the case of isotype control antibodies. The above results suggest a strong association between nanoparticles and SCARB1 which regulate cholesterol transfer across the receptor. Furthermore, in SUDHL4 cells, HDL NPs diminished total cholesterol in a dose-dependent manner (Figure 4E). Changes of the cholesterol biosynthetic gene expression after HDL NPs or human HDL treatment in SUHDL4, TMD8, and HBL-1 cell lines were also investigated. It was found that, with the exposure of HDL NPs, cholesterol biosynthetic gene DHCR7 was strongly elevated in SUHDL4 cells (Figure 4F). The combination of HDL-NPs and ibrutinib exhibited a synergistic effect, leading to a significant reduction in the total cholesterol levels of both TMD8 and HBL-1 cells. In vivo experiment showed that upon HDL NPs treatment, the tumor volumes of SUDHL4 xenografts SCID-beige mice were significantly reduced (Figure 4G). These results demonstrated the feasibility of cholesterol depleting strategy as a therapeutic intervention for lymphoma.
 |
Figure 4 (A) Schematic illustrating the effect of HDL NPs on Diffuse Large B-Cell Lymphoma. (B) HDL NPs (solid line) induce cell death in the GC DLBCL cell line SUDHL4, but human HDL (dotted line) does not induce lymphoma cell death. *p < 0.05 vs PBS control. (C and D) HDL NPs efflux cholesterol from SUDHL4 (left) and TMD8 (right) cells. *p < 0.05 vs no antibody. (E) Total cell cholesterol content treated with PBS or HDL NPs. *p < 0.05 vs 0 nM HDL NP. (F) HDL NPs modulate cholesterol biosynthesis gene expression. *p < 0.05 vs PBS control. (G) Tumor growth of SUDHL4 tumors after different treatments. *p < 0.0001 vs all other treatment groups by one-way ANOVA. Tp < 0.0001 vs PBS control only by one-way ANOVA. Abbreviations: SCARB1, scavenger receptor type B1; HDL, high-density lipoprotein; DLBCL, diffuse large B-cell lymphoma. Notes: Rink, JS, Yang S, Cen O, et al. Rational Targeting of Cellular Cholesterol in Diffuse Large B-Cell Lymphoma (DLBCL) Enabled by Functional Lipoprotein Nanoparticles: A Therapeutic Strategy Dependent on Cell of Origin. Mol Pharm. 2017; 14(11): 4042–4051. Copyright 2017 American Chemical Society.76 |
In another study, Kuai et al constructed synthetic HDL (sHDL) nanodisks with a tumor-targeting effect.75 In their study, sHDL nanodisks were prepared with ApoA1 mimetic peptide (22A) in combination with phospholipid. These nanodisks were then loaded with a novel semisynthetic compound, withalongolide A 4,19,27-triacetate (WGA-TA), which demonstrated highly effective antitumor activity.99 The cytotoxicity experiments showed that blank-sHDL possessed an antitumor effect in the H295R cell line which expressed a high level of SCARB1. Such effect was attributed to sHDL-mediated regulation of cholesterol efflux and the inhibition of corticosteroid synthesis. In addition, the SCARB1 antibody downregulated WGA-TA-sHDL regulation in H295 cells. Finally, the research group verified the WGA-TA-sHDL nanodisk-mediated suppression of ACC growth in H295R xenografts in vivo. Based on the above results, the tumor volume in WGA-TA-sHDL-treated group was substantially reduced relative to the other groups, thereby suggesting a major benefit of employing sHDL for WGA-TA in cancer treatment.
Cholesterol Metabolism Interference
Interference with Cholesterol Esterification
In addition to obtaining large amounts of cholesterol from de novo biosynthetic pathway and increased uptake, the anomalous metabolic requirements drive cancer cells to convert excess cholesterol to less toxic cholesterol esters (CE) via acyl-CoA cholesterol acyltransferase-1 (ACAT-1) in the endoplasmic reticulum and reserve them as cholesteryl ester transfer protein-mediated lipid droplets.24 Cholesteryl ester transfer protein knockout breast cancer cells have been reported to pose less resistance toward cytotoxic compounds.100 ACAT-1 is widely expressed in most tissues and overexpressed in tumors like pancreatic cancer, glioblastoma, and prostate cancer.22 CE serves as a cholesterol reservoir, and cancer cells use it via a swift transition between CE and free cholesterol when demand increases. Besides, CE offers considerable advantages to cancer cells when they encounter adverse stimuli like drug therapy.6 Inhibition of ACAT1 reduces CE levels and dampens cell proliferation and aggressiveness of tumors.
In one study, a kind of systemically injectable nanodrug was developed to encapsulate avasimibe (an ACAT-1 inhibitor) within human serum albumin aiming at blocking cholesterol esterification in tumors.83 Reduced CE packing in lipid droplets and enhanced intracellular free cholesterol contents appear to stiffen the endoplasmic reticulum membrane, thus leading to apoptosis and proliferation inhibition in human prostate, pancreatic, lung, and colon cancer cells. The accumulation of nanodrugs in tumor tissue was 4 times higher than the half maximal inhibitory concentration evidenced by the significantly suppressed tumor growth.
Targeted CE also reinvigorates the cancer-suppressing effect of CD8+ T cells. Unfortunately, T cell metabolism is often regulated by tumor cells, thus weakening the antitumor response. Elevating the membranal cholesterol levels promotes T-cell antigen receptor accumulation and activates CD8+ T cells.101 Liu et al developed an intelligent liposome for codelivery of avasimibe and photosensitizers.102 Blocking cholesterol esterification resulted in increased cholesterol levels in B16-F10 cells, downregulated SREBP2, decreased membrane cholesterol and subsequently dampened membrane cholesterol-dependent integrin-induced migration. Similarly, blocking cholesterol metabolism reduced cholesterol levels in T cells’ plasma membranes and reinvigorated T cells. Results demonstrated that nanovesicles coupled with irradiation showed great potential to induce ICDs to generate adaptive antigens in the in vivo and in vivo studies. The infiltration of activated phenotype of TNFα+ CD8+ T cells in TME was increased in B16-F10 mouse tumor model. As a result, the cholesterol metabolism and photodynamic cancer immunotherapy significantly delayed the growth of tumors and prolonged the survival rate of animals. However, it was reported that excess cholesterol in TME promotes CD8+ T cell exhaustion via the endoplasmic reticulum stress-XBP1 network.68 The effect of cholesterol on T-cell function seems controversial. Therefore, understanding the specific role of cholesterol in various immune cells within different tumor types will aid in the development of more precise therapies.
Promotion of Membrane Cholesterol Oxidation
Elevated cholesterol is a prerequisite for the rapid proliferation of cancer cells to achieve membrane biogenesis and other functional requirements.6 LRs are densely populated cholesterol- and sphingolipid-enrich domains within the plasma membrane and are involved in cancer proliferation, adhesion, migration, and drug resistance52 as described in Cholesterol-Related Tumorigenesis And Progression of this paper. Cholesterol regulates LR signaling, increases membrane ordering, and reduces membrane fluidity and permeability.46,103 Multidrug resistance (MDR) cells display increased cholesterol content and decreased membrane fluidity for antagonizing drug penetration.56 Further, MDR efflux transporters, members of the ATP-binding cassette superfamily, reside in LR, contributing to drug efflux. ABC in LR regulates intracellular cholesterol homeostasis, whereas, in turn, cholesterol influences ABC transporter content, recycling, and physiological activity.24,56 Based on the above analysis, cholesterol consumption is expected to improve the mobility of MDR plasma membranes and potentially reverse drug resistance.
In 2022, Du et al designed a nanosystem with cholesterol cascade catalytic consumption capacity.87 This system involved immobilizing cholesterol oxidase (COD) onto a metal–organic framework (MOF) carrier to facilitate cholesterol consumption. The nanosystem was further modified with a chondroitin sulfate gel shell for CD44 targeting (Figure 5A). To investigate the cholesterol consumption capacity of the nanosystem and evaluate its influence on plasma membrane fluidity, MβCD was applied as a positive control agent. Results showed that the DOX@MOF-COD@CS group significantly reduced the cholesterol content of drug-resistant cells (Figure 5B) and enhanced plasma membrane fluidity similar to the function of MβCD (Figure 5C). Meanwhile, the confocal laser microscope results showed slight green fluorescence in drug-treated groups, suggesting that cholesterol consumption strongly influences LR production within the plasma membrane (Figure 5D). As depicted in Figure 5E, the MOF cells catalyzed the formation of hydroxyl radicals from intracellular H2O2, thus, emitting green fluorescence. Moreover, stronger green fluorescence was seen in the DOX@MOF-COD@CS group, suggesting that COD could release additional H2O2. In vivo antitumor experiment revealed that the DOX@MOF-COD@CS group reversed tumor doxorubicin resistance and had the best antitumor effect (Figure 5F). Elsewhere, a dual drug (COD and DOX)-loaded tumor cell membrane-coated metal–organic framework was designed for drug-resistant tumor therapy. The researchers demonstrated that COD catalyzed the oxidative metabolism of cholesterol leading to the production of H2O2. Then, H2O2 would be catalytically decomposed by metal–organic framework to form hydroxyl radicals. Therefore, the designed nanodrug could effectively treat drug-resistant breast cancer.86 Taking cholesterol in drug resistance cell membranes as a “breakthrough”, combined with the cholesterol-related anti-apoptotic pathway in the drug resistance mechanism, the above studies will provide a novel sense for reversing multidrug resistance of tumors.
 |
Figure 5 (A) Schematic illustrating the effect of DOX@MOF-COD@CS nanoparticles on multidrug resistant tumor. (B) Cholesterol content in MCF-7/ADR cells. (C) The plasma membrane fluidity in MCF-7/ADR cells. (D) The formation of membrane lipid rafts in MCF-7/ADR cell. (E) Reactive oxygen species detection in MCF-7/ADR cell. (F) Tumor growth of MCF-7/ADR tumors after different treatments. **p < 0.01, ***p < 0.001, and n.s. = no significant difference. Abbreviations: DOX@MOF-COD@CS, doxorubicin loaded nanosystem with a cholesterol cascade catalytic consumption; CS, chondroitin sulfate gel. Notes: Reproduced from Du, B, Zheng M, Ma HZ, et al. Nanozyme-natural enzymes cascade catalyze cholesterol consumption and reverse cancer multidrug resistance. Journal of nanobiotechnology. 2022; 20(1): 209–223. Creative Commons.87 |
Cholesterol Depletion
In addition to inhibiting the synthesis of cholesterol, interfering with cellular uptake, promoting its biological metabolism, or extracting cholesterol from the cell membrane could be another potential strategy to disrupt the structure of tumor plasma membrane. MβCD is utilized to deplete cholesterol by forming a complex (host-guest interaction) with cholesterol extracted from the plasma membrane.104 It is reported that MβCD decreased membrane cholesterol by disrupting LR and induced apoptosis by disrupting signal transduction between phosphoinositide 3-kinases and protein kinase B.105,106 The combination of MβCD and tamoxifen was employed for the treatment of melanoma. MβCD was used to lower cholesterol levels and enhance the cell sensitivity to tamoxifen, resulting in a 75% decrease in tumor weight.107 All of these results suggest that MβCD is a potential antitumor drug. Song et al designed a nano-assembly (NA) targeting the CD44 receptor, which effectively damaged the structure of LR as a potential approach for breast cancer therapy.81 In their study, MβCD was ligated to a hyaluronic acid-ceramide (HACE) structure through an ester linkage. Hyaluronic acid (HA) was employed to target tumor cells due to its binding affinity with CD44, which is often overexpressed in these cells (Figure 6A). Anticancer potentials of HACE-MbCD NA were investigated, showing that MDA-MB-231 cells displayed reduced cell proliferation and enhanced cell apoptosis (Figure 6B and C). Filipin III selectively binds to free plasma membrane cholesterol. Therefore, filipin III staining was used to assess the cholesterol efflux from the plasma membrane by MβCD. HACE-MbCD NA significantly increased cholesterol outflow through the plasma membrane (Figure 6D). When tested in the animal model, HACE-MbCD NA showed the strongest antitumor effect than other treatments (Figure 6E). All these findings suggested that CD44 receptor-targeted HACE-MbCD NA, retaining cholesterol depletion activity from cancer cells, might be one of remarkable nanosystems for breast cancer therapy. Erlotinib received approval for clinical applications in non-small cell lung cancer. In another study, Gamze et al designed a cholesterol-deleting cyclodextrin nanoparticle that encapsulated erlotinib to combat drug-resistant cancer.82 The results showed that nanoplatforms were able to extract membrane cholesterol to overcome drug resistance and improve antitumor efficacy.
 |
Figure 6 (A) Schematic illustrating the effect of HACE-MbCD NA on MDA-MB-231 tumor. (B) Antiproliferation assay of MbCD and HACE-MbCD in NIH3T3, HUVEC, and MDA-MB-231 cells. (C) Apoptotic efficacies of HACE-MbCD NA in MDA-MB-231 cells. *p < 0.05 vs the control group. &p < 0.05 vs the MbCD group. #p < 0.05 vs the HACE NA group. (D) Cholesterol capture assay of HACE-MbCD NA in MDA-MB-231 cells. (E) Tumor growth of MDA-MB-231 tumors after different treatments. *p < 0.05 vs the control group. #p < 0.05 vs the HACE NA group. &p < 0.05 vs the MbCD group. Abbreviations: HACE-MbCD, hyaluronic acid-ceramide- methyl-β-cyclodextrin nanoassembly; HA, hyaluronic acid; DOX, doxorubicin. Notes: Lee, SY, Ko SH, Shim JS, et al. Tumor Targeting and Lipid Rafts Disrupting Hyaluronic Acid-Cyclodextrin-Based Nanoassembled Structure for Cancer Therapy. ACS applied materials & interfaces. 2018; 10(43): 36628–36640. Copyright 2018 American Chemical Society.81 |
Combination Treatment
The regulation of cholesterol homeostasis is complex. Targeting only one pathway of cholesterol metabolism may have limited anticancer effects, as it may not account for feedback loops or activation of other pro-oncogenic pathways. Nanoplatform can achieve combined effects in cholesterol synthesis, absorption, and esterification. Meanwhile, cholesterol regulating strategies can work in synergy with other strategies, such as chemotherapy and immunotherapy, to enhance their anticancer impact. Nanoplatforms offer an avenue for loading drugs with different therapeutic mechanisms and delivering them to tumor sites. After verifying the combined therapeutic effect of metformin and atorvastatin in a clinical study (NCT01980823), constructing tumor-targeted nanoplatforms can further improve the concentration of drugs in tumor sites and reduce side effects. Another clinical trial (NCT04698941) applied the combined usage of paclitaxel and simvastatin against non-small cell lung cancer. The following studies can integrate cholesterol modulators into commercially available paclitaxel nanoformulations (such as Cynviloq®, Abraxane®, and Lipusu®) to achieve nanocarrier-mediated combination therapy of tumors. Combination therapeutic nanodrugs developed in this way will be more likely to achieve clinical translation.
Nanoplatforms combining cholesterol metabolism inhibitors and chemotherapy enable the reversal of drug resistance. For instance, Jin et al developed legumain-activatable liposomes containing the cholesterol synthesis inhibitor (SV) and PTX. When administered intravenously, the liposomes could deliver SV and PTX to the tumor site through the EPR effect. Subsequently, the liposome released the SV to disrupt the structure of lipid raft and suppress the expression of integrin-β3, leading to inhibition of the focal adhesion kinase signaling pathway and re-sensitizing the drug-resistant cancer cells to PTX. In this study, the developed nanoplatform achieved co-delivery and specific release of two drugs to the tumor site, thereby providing synergistic tumor treatment.61
As stated before, HDL nanoparticles as a carrier not only increase the accumulation of cholesterol in TME via SCARB1 but also regulate cholesterol homeostasis in the plasma membrane. A parenteral SV-loaded HDL nanoformulation consisting of DPPC and ApoA-1 mimicking peptide was developed and combined with radiotherapy for potent anticancer effects.89 The researchers found that the effluxed cholesterol content was substantially enhanced (137.8 ± 18.1% vs non-treated cells) after being treated with the HDL nanoformulations for 5 h. The enhanced SV intracellular level and HDL-induced cholesterol efflux may confer radiosensitization of NPs equivalent to 10-fold and 5-fold doses of free SV in 2D and 3D cultures. In vivo experiments also showed that the mean tumor size in the simvastatin-HDL NPs and radiation combination group was the smallest among all treated and control groups, indicating the excellent collaborative therapy effect of HDL nanoformulations. HDL NPs and SV regulate cholesterol homeostasis in the cell membrane. Moreover, the membrane cholesterol content altered the radiation sensitivity of cancer cells and cellular signaling related to radiation-induced immune responses, thus achieving synergistic anti-tumor effects.
Nanoplatforms for combination treatments targeting cholesterol metabolism hold great promise in clinical cancer therapy. For instance, nanoplatforms facilitate the implementation of personalized medicine that is customized to each patient. By developing nanoplatforms tailored to meet individual patients’ tumor types, genetic profiles and disease progression, patients can minimize side effects and maximize efficacy during treatment. Advances in specific research fields may accelerate the application of nanoplatforms in combination therapies targeting cholesterol metabolism in cancer. Firstly, future studies should expand our understanding of the mechanisms underlying cancer-related cholesterol metabolism, particularly its role in drug resistance. Secondly, researchers should design co-delivery technologies for multiple drugs harboring different properties and improve targeting delivery efficiency to boost the clinical application of combination therapy nanoplatforms. Furthermore, the dose-effect relationship, release sequence/cycle, and pharmacokinetic studies of multiple drugs in combined therapy nanoplatforms need to be elucidated in preclinical studies, rather than simply mixing different drugs in a random ratio.
Summary and Prospects
This paper demonstrates the role of cholesterol in tumorigenesis and provides an overview of cholesterol-integrated membrane lipid nanotherapeutics for cancer therapy. As epidemiological studies have shown the correlation between high cholesterol expression and tumor development, a large number of clinical studies on the combination of cholesterol-lowering drugs (like statins) with chemotherapy drugs have sprung up worldwide (Table 1). Although combined tumor treatments have shown promising advances in clinical trials, current clinical studies have primarily focused on inhibiting cholesterol synthesis in tumor cells, which may be a relatively limited approach. Building on the insights presented in this article, future research should aim to reveal novel therapeutic options and treatment strategies. This includes exploring avenues like cholesterol uptake, transport, metabolism, and the blockade of cholesterol-related signaling pathways as promising targets for drug development. Regarding nanodrugs designed to disrupt cholesterol synthesis in tumor cells, safe and functional materials (like high-density lipoprotein) or organic molecules (like MβCD) should be created to improve patient management. Incorporation of cholesterol metabolism interfering agents into commercially available nanodrugs may be a promising strategy for clinical translation.
Despite substantial improvements in cholesterol regulation tumor nanotherapeutics, the clinical applications of these approaches are still constrained by several challenges. Firstly, cholesterol-modulating therapy strategies have only been applied to a few cancer types, such as breast and prostate cancer. Therefore, before implementing a therapeutic strategy based on cholesterol regulation, it is necessary to elucidate the degree of cholesterol expression and its related roles in different types of tumors. The application of high-throughput siRNA screen technology in screening cholesterol-related genes in cancer patients could achieve personalized treatment of tumors. Secondly, a majority of the aforementioned nanotherapeutics focused on assessing curative efficacy rather than safety. The safety of tumor nanotherapeutics at the organismal level demands more comprehensive investigations. Moreover, further studies are warranted to examine the downstream outcomes of cholesterol depletion intervention, involving different signaling pathways. It is also important to gain a better understanding of potential side effects and the long-term therapeutic outcomes associated with these interventions. Lastly, the industrial feasibility and economic aspects of membranal cholesterol-regulating nanoformulations should also be investigated, such as simple prescription, controllable cost, easy transportation, and long-term stability. Developing a simple yet effective approach in this regard is likely to facilitate the clinical translation of these therapies. Thus, we recommend additional mechanistic investigations involving cancer-related cholesterol metabolism to augment future cancer therapies. In general, the content described here is only the tip of the iceberg in this field. We expect that further research will increasingly concentrate on cholesterol-integrated tumor therapeutic strategies to advance toward their clinical applications.
Abbreviations
27-HC, 27-hydroxycholesterol; ACAT-1, acyl-CoA cholesterol acyltransferase-1; ATP, adenosine triphosphate; CE, cholesterol esters; COD, cholesterol oxidase; DLBCL, diffuse large B-cell lymphoma; DOX, doxorubicin; DOX@MOF-COD@CS, DOX loaded nanosystem with a cholesterol cascade catalytic consumption; EALP, matrix metalloproteinase-2 -sensitive tumor-penetrable nanovesicle; EMT, epithelial-mesenchymal transition; FDPS, farnesyl diphosphate synthase; HA, hyaluronic acid; HACE, hyaluronic acid-ceramide; HACE-MbCD NA, hyaluronic acid-ceramide- methyl-β-cyclodextrin nanoassembly; HDL, high-density lipoprotein; HDL NPs, high-density lipoprotein nanoparticles; HMGCR, hydroxy-3-methylglutaryl-CoA reductase; ICIs, immune checkpoint inhibitors; LR, lipid raft; LXR, liver X receptor; MDR, multidrug resistance; MOF, metal–organic framework; mTORC1, mammalian target of rapamycin C1; MβCD/MbCD, methyl-β-cyclodextrin; NPC1, niemann-pick C1 protein; p-gp, p-glycoprotein; PTX, paclitaxel; SCARB1, scavenger receptor type B1; Smo, smoothened; SREBP2, sterol regulatory element-binding protein 2; STARD3, steroidogenic acute regulatory related lipid transfer domain-3; SV, simvastatin; TAMs, tumor-associated macrophages; TME, tumor microenvironment; WGA-TA-sHDL, withalongolide A 4,19,27-triacetate loaded synthetic HDL nanodisks.
Funding
This study was financially supported by the National Natural Science Foundation of China (82103505 and 82071616), the Zhejiang Provincial Natural Science Foundation Public Welfare Projects of China (LTGC23H050001), and the Medical Health Science and Technology Project of Zhejiang Provincial Health Commission (2021RC005).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol. 2008;9(2):125–138. doi:10.1038/nrm2336
2. Murai T. Cholesterol lowering: role in cancer prevention and treatment. Biol Chem. 2015;396(1):1–11. doi:10.1515/hsz-2014-0194
3. Smith B, Land H. Anticancer activity of the cholesterol exporter ABCA1 gene. Cell Rep. 2012;2(3):580–590. doi:10.1016/j.celrep.2012.08.011
4. Krycer JR, Brown AJ. Cholesterol accumulation in prostate cancer: a classic observation from a modern perspective. Biochim Biophys Acta Rev Cancer. 2013;1835(2):219–229. doi:10.1016/j.bbcan.2013.01.002
5. Llaverias G, Danilo C, Mercier I, et al. Role of cholesterol in the development and progression of breast cancer. Am J Pathol. 2011;178(1):402–412. doi:10.1016/j.ajpath.2010.11.005
6. Huang B, Song BL, Xu C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab. 2020;2(2):132–141. doi:10.1038/s42255-020-0174-0
7. Shafique K, McLoone P, Qureshi K, et al. Cholesterol and the risk of grade-specific prostate cancer incidence: evidence from two large prospective cohort studies with up to 37 years’ follow up. Bmc Cancer. 2012;12(25):25–33. doi:10.1186/1471-2407-12-25
8. Pelton K, Freeman MR, Solomon KR. Cholesterol and prostate cancer. Curr Opin Pharmacol. 2012;12(6):751–759. doi:10.1016/j.coph.2012.07.006
9. Allott EH, Howard LE, Cooperberg MR, et al. Serum lipid profile and risk of prostate cancer recurrence: results from the SEARCH database. Cancer Epidemiol Biomarkers Prev. 2014;23(11):2349–2356. doi:10.1158/1055-9965.Epi-14-0458
10. Kuzu OF, Noory MA, Robertson GP. The role of cholesterol in cancer. Cancer Res. 2016;76(8):2063–2070. doi:10.1158/0008-5472.Can-15-2613
11. Chimento A, Casaburi I, Avena P, et al. Cholesterol and its metabolites in tumor growth: therapeutic potential of statins in cancer treatment. Front Endocrinol. 2018;9:807. doi:10.3389/fendo.2018.00807
12. Ding X, Zhang WH, Li S, et al. The role of cholesterol metabolism in cancer. Am J Can Res. 2019;9(2):219–227.
13. Costa GA, de Souza SB, da Silva Teixeira LR, et al. Tumor cell cholesterol depletion and V-ATPase inhibition as an inhibitory mechanism to prevent cell migration and invasiveness in melanoma. Biochim Biophys Acta Gen Subj. 2018;1862(3):684–691. doi:10.1016/j.bbagen.2017.12.006
14. Lyu J, Yang EJ, Head SA, et al. Pharmacological blockade of cholesterol trafficking by cepharanthine in endothelial cells suppresses angiogenesis and tumor growth. Cancer Lett. 2017;409:91–103. doi:10.1016/j.canlet.2017.09.009
15. Wen YA, Xiong X, Zaytseva YY, et al. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. 2018;9(3):265. doi:10.1038/s41419-018-0330-6
16. Wei X, Song M, Jin G, et al.. Multidimensional profiling of functionalized photothermal nanoplatforms for synergistic cancer immunotherapy: design, strategy, and challenge. Coord Chem Rev. 2024:499. doi:10.1016/j.ccr.2023.215488
17. Wei X, Song M, Jiang G, et al. Progress in advanced nanotherapeutics for enhanced photodynamic immunotherapy of tumor. Theranostics. 2022;12(12):5272–5298. doi:10.7150/thno.73566
18. Wei X, Wang J, Liang M, et al. Development of functional nanomedicines for tumor associated macrophages-focused cancer immunotherapy. Theranostics. 2022;12(18):7821–7852. doi:10.7150/thno.78572
19. Lan J. Overview of Application of Nanomaterials in Medical Domain. Contrast Media Mol Imaging. 2022;2022:3507383. doi:10.1155/2022/3507383
20. Weidong Fei JY, Xiaodong W, Yang S, et al.. Perturbing plasma membrane lipid: a new paradigm for tumor nanotherapeutics. Theranostics. 2023;13(8):2471–2491. doi:10.7150/thno.82189
21. Wang J, Zhu M, Nie G. Biomembrane-based nanostructures for cancer targeting and therapy: from synthetic liposomes to natural biomembranes and membrane-vesicles. Adv Drug Delivery Rev. 2021;178:113974. doi:10.1016/j.addr.2021.113974
22. Xu H, Zhou S, Tang Q, et al. Cholesterol metabolism: new functions and therapeutic approaches in cancer. BBA. 2020;1874(1):188394. doi:10.1016/j.bbcan.2020.188394
23. Wang Y, Rogers PM, Su C, et al. Regulation of cholesterologenesis by the oxysterol receptor, LXR alpha. J Biol Chem. 2008;283(39):26332–26339. doi:10.1074/jbc.M804808200
24. Gu L, Saha ST, Thomas J, et al. Targeting cellular cholesterol for anticancer therapy. FEBS J. 2019;286(21):4192–4208. doi:10.1111/febs.15018
25. Guixà-González R, Albasanz JL, Rodriguez-Espigares I, et al. Membrane cholesterol access into a G-protein-coupled receptor. Nat Commun. 2017;8(1):14505. doi:10.1038/ncomms14505
26. Kiskova T, Kassayova M. Resveratrol action on lipid metabolism in cancer. Int J Mol Sci. 2019;20(11):2704. doi:10.3390/ijms20112704
27. Zhao C, Dahlman-Wright K. Liver X receptor in cholesterol metabolism. J Endocrinol. 2010;204(3):233–240. doi:10.1677/JOE-09-0271
28. Vedin LL, Lewandowski SA, Parini P, et al. The oxysterol receptor LXR inhibits proliferation of human breast cancer cells. Carcinogenesis. 2009;30(4):575–579. doi:10.1093/carcin/bgp029
29. Codini M, Garcia-Gil M, Albi E. Cholesterol and sphingolipid enriched lipid rafts as therapeutic targets in cancer. Int J Mol Sci. 2021;22(2):726. doi:10.3390/ijms22020726
30. Jaffres P-A, Gajate C, Bouchet AM, et al. Alkyl ether lipids, ion channels and lipid raft reorganization in cancer therapy. Pharmacol Ther. 2016;165:114–131. doi:10.1016/j.pharmthera.2016.06.003
31. Mollinedo F, Gajate C. Lipid rafts as signaling hubs in cancer cell survival/death and invasion: implications in tumor progression and therapy: thematic review series: biology of lipid rafts. J Lipid Res. 2020;61(5):611–635. doi:10.1194/jlr.TR119000439
32. Wu Z, Li X, Guo D, et al. Lipid raft-associated PI3K/Akt/SREBP1 signaling regulates coxsackievirus A16 (CA16) replication. Vet Microbiol. 2021;252:108921. doi:10.1016/j.vetmic.2020.108921
33. Zeng J, Zhang H, Tan Y, et al. Aggregation of lipid rafts activates c-met and c-Src in non-small cell lung cancer cells. BMC Cancer. 2018;18:1–11. doi:10.1186/s12885-018-4501-8
34. Zhuang L, Kim J, Adam RM, et al. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J Clin Invest. 2005;115(4):959–968. doi:10.1172/JCI200519935
35. Solomon KR, Freeman MR. Do the cholesterol-lowering properties of statins affect cancer risk?. Trends Endocrinol Metab. 2008;19(4):113–121. doi:10.1016/j.tem.2007.12.004
36. Meng Y, Heybrock S, Neculai D, et al. Cholesterol handling in lysosomes and beyond. Trends Cell Biol. 2020;30(6):452–466. doi:10.1016/j.tcb.2020.02.007
37. Thelen AM, Zoncu R. Emerging roles for the lysosome in lipid metabolism. Trends Cell Biol. 2017;27(11):833–850. doi:10.1016/j.tcb.2017.07.006
38. Meneses-Salas E, García-Melero A, Kanerva K, et al. Annexin A6 modulates TBC1D15/Rab7/StARD3 axis to control endosomal cholesterol export in NPC1 cells. Cell Mol Life Sci. 2020;77:2839–2857. doi:10.1007/s00018-019-03330-y
39. Vassilev B, Sihto H, Li S, et al. Elevated levels of StAR-related lipid transfer protein 3 alter cholesterol balance and adhesiveness of breast cancer cells: potential mechanisms contributing to progression of HER2-positive breast cancers. Am J Pathol. 2015;185(4):987–1000. doi:10.1016/j.ajpath.2014.12.018
40. Huang Y, Li C, Zhang X, et al. Nanotechnology-integrated ovarian cancer metastasis therapy: insights from the metastatic mechanisms into administration routes and therapy strategies. Int J Pharm. 2023;636:122827–122843. doi:10.1016/j.ijpharm.2023.122827
41. Lesley J, Hyman R, Kincade PW. CD44 and its interaction with extracellular matrix. Advan Immunol. 1993;54:271–335. doi:10.1016/S0065-2776(08)60537-4
42. Murai T, Maruyama Y, Mio K, et al. Low cholesterol triggers membrane microdomain-dependent CD44 shedding and suppresses tumor cell migration. J Biol Chem. 2011;286(3):1999–2007. doi:10.1074/jbc.M110.184010
43. McDonnell DP, Park S, Goulet MT, et al. Obesity, cholesterol metabolism, and breast cancer pathogenesis. Cancer Res. 2014;74(18):4976–4982. doi:10.1158/0008-5472.Can-14-1756
44. Warita K, Warita T, Beckwitt CH, et al. Statin-induced mevalonate pathway inhibition attenuates the growth of mesenchymal-like cancer cells that lack functional E-cadherin mediated cell cohesion. Sci Rep. 2014;4:7593–7600. doi:10.1038/srep07593
45. Hendrich AB, Michalak K. Lipids as a target for drugs modulating multidrug resistance of cancer cells. Curr Drug Targets. 2003;4(1):23–30. doi:10.2174/1389450033347172
46. Peetla C, Vijayaraghavalu S, Labhasetwar V. Biophysics of cell membrane lipids in cancer drug resistance: implications for drug transport and drug delivery with nanoparticles. Adv Drug Delivery Rev. 2013;65(13–14):1686–1698. doi:10.1016/j.addr.2013.09.004
47. Cohen AW, Hnasko R, Schubert W, et al. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004;84(4):1341–1379. doi:10.1152/physrev.00046.2003
48. Eytan GD, Regev R, Oren G, et al. The role of passive transbilayer drug movement in multidrug resistance and its modulation. J Biol Chem. 1996;271(22):12897–12902. doi:10.1074/jbc.271.22.12897
49. Preetha A, Banerjee R, Huilgol N. Tensiometric profiles and their modulation by cholesterol: implications in cervical cancer. Cancer Invest. 2007;25(3):172–181. doi:10.1080/07357900701209053
50. Lajoie P, Nabi IR. Lipid rafts, caveolae, and their endocytosis. Internat Rev Cel Molecul Biol. 2010;282:135–163. doi:10.1016/S1937-6448(10)82003-9
51. Ye DM, Ye SC, Yu SQ, et al. Drug-resistance reversal in colorectal cancer cells by destruction of flotillins, the key lipid rafts proteins. Neoplasma. 2019;66(4):576–583. doi:10.4149/neo_2018_180820N633
52. Zalba S, Ten Hagen TL. Cell membrane modulation as adjuvant in cancer therapy. Cancer Treat Rev. 2017;52:48–57. doi:10.1016/j.ctrv.2016.10.008
53. Chantemargue B, Di Meo F, Berka K, et al. Structural patterns of the human ABCC4/MRP4 exporter in lipid bilayers rationalize clinically observed polymorphisms. Pharmacol Res. 2018;133:318–327. doi:10.1016/j.phrs.2018.02.029
54. Meyer Dos Santos S, Weber CC, Franke C, et al. Cholesterol: coupling between membrane microenvironment and ABC transporter activity. Biochem Biophys Res Commun. 2007;354(1):216–221. doi:10.1016/j.bbrc.2006.12.202
55. Raghavan V, Vijayaraghavalu S, Peetla C, et al. Sustained epigenetic drug delivery depletes cholesterol-sphingomyelin rafts from resistant breast cancer cells, influencing biophysical characteristics of membrane lipids. Langmuir. 2015;31(42):11564–11573. doi:10.1021/acs.langmuir.5b02601
56. Kopecka J, Trouillas P, Gasparovic AC, et al. Phospholipids and cholesterol: inducers of cancer multidrug resistance and therapeutic targets. Drug Resist Updates. 2020;49:100670–100740. doi:10.1016/j.drup.2019.100670
57. Kim HY, Kim DK, Bae SH, et al. Farnesyl diphosphate synthase is important for the maintenance of glioblastoma stemness. Exp Mol Med. 2018;50(10):1–12. doi:10.1038/s12276-018-0166-2
58. Kim S, Lee M, Dhanasekaran DN, et al. Activation of LXRɑ/β by cholesterol in malignant ascites promotes chemoresistance in ovarian cancer. BMC Cancer. 2018;18(1):1232. doi:10.1186/s12885-018-5152-5
59. Kopecka J, Porto S, Lusa S, et al. Zoledronic acid-encapsulating self-assembling nanoparticles and doxorubicin: a combinatorial approach to overcome simultaneously chemoresistance and immunoresistance in breast tumors. Oncotarget. 2016;7(15):20753–20772. doi:10.18632/oncotarget.8012
60. Henriques Palma GB, Kaur M. Cholesterol depletion modulates drug resistance pathways to sensitize resistant breast cancer cells to tamoxifen. Anticancer Res. 2022;42(1):565–579. doi:10.21873/anticanres.15514
61. Jin H, He Y, Zhao P, et al. Targeting lipid metabolism to overcome EMT-associated drug resistance via integrin beta3/FAK pathway and tumor-associated macrophage repolarization using legumain-activatable delivery. Theranostics. 2019;9(1):265–278. doi:10.7150/thno.27246
62. Zou Y, Yu X, Zhou C, et al. Adverse effects of low serum lipoprotein cholesterol on the immune microenvironment in gastric cancer: a case‒control study. Lipids Health Dis. 2022;21(1):150–164. doi:10.1186/s12944-022-01766-z
63. Hao M, Hou S, Li W, et al. Combination of metabolic intervention and T cell therapy enhances solid tumor immunotherapy. Sci, trans med. 2020;12(571):6667–6686. doi:10.1126/scitranslmed.aaz6667
64. Liu C, Yang XV, Wu J, et al. Oxysterols direct B-cell migration through EBI2. Nature. 2011;475(7357):519–523. doi:10.1038/nature10226
65. Liu X, Bao X, Hu M, et al. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature. 2020;588(7839):693–698. doi:10.1038/s41586-020-2911-7
66. Wang R, Liu Z, Fan Z, et al. Lipid metabolism reprogramming of CD8(+) T cell and therapeutic implications in cancer. Cancer Lett. 2023;567:216267–216309. doi:10.1016/j.canlet.2023.216267
67. Liu X, Hartman CL, Li L, et al. Reprogramming lipid metabolism prevents effector T cell senescence and enhances tumor immunotherapy. Sci, trans med. 2021;13(587):6314–6334. doi:10.1126/scitranslmed.aaz6314
68. Ma XZ, Bi EG, Lu Y, et al. Cholesterol Induces CD8(+) T cell exhaustion in the tumor microenvironment. Cell Metab. 2019;30(1):143–162. doi:10.1016/j.cmet.2019.04.002
69. Lim SA, Su W, Chapman NM, et al. Lipid metabolism in T cell signaling and function. Nat Chem Biol. 2022;18(5):470–481. doi:10.1038/s41589-022-01017-3
70. Baek AE, Yu YA, He S, et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat Commun. 2017;8(1):864–874. doi:10.1038/s41467-017-00910-z
71. Goossens P, Rodriguez-Vita J, Etzerodt A, et al. Membrane cholesterol efflux drives tumor-associated macrophage reprogramming and tumor progression. Cell Metab. 2019;29(6):1376–1389. doi:10.1016/j.cmet.2019.02.016
72. Shi J, Kantoff PW, Wooster R, et al. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17(1):20–37. doi:10.1038/nrc.2016.108
73. Wei X, Song M, Li W, et al. Multifunctional nanoplatforms co-delivering combinatorial dual-drug for eliminating cancer multidrug resistance. Theranostics. 2021;11(13):6334–6354. doi:10.7150/thno.59342
74. Yang S, Damiano MG, Zhang H, et al. Biomimetic, synthetic HDL nanostructures for lymphoma. Proc Natl Acad Sci USA. 2013;110(7):2511–2516. doi:10.1073/pnas.1213657110
75. Kuai R, Subramanian C, White PT, et al. Synthetic high-density lipoprotein nanodisks for targeted withalongolide delivery to adrenocortical carcinoma. Int j Nanomed. 2017;12:6581–6594. doi:10.2147/ijn.s140591
76. Rink JS, Yang S, Cen O, et al. Rational targeting of cellular cholesterol in diffuse large B-Cell Lymphoma (DLBCL) enabled by functional lipoprotein nanoparticles: a therapeutic strategy dependent on cell of origin. Mol Pharm. 2017;14(11):4042–4051. doi:10.1021/acs.molpharmaceut.7b00710
77. Subramanian C, Kuai R, Zhu Q, et al. Synthetic high-density lipoprotein nanoparticles: a novel therapeutic strategy for adrenocortical carcinomas. Surgery. 2016;159(1):284–294. doi:10.1016/j.surg.2015.08.023
78. Rink JS, Lin AY, McMahon KM, et al.. Targeted reduction of cholesterol uptake in cholesterol-addicted lymphoma cells blocks turnover of oxidized lipids to cause ferroptosis. J Biol Chem. 2021:296. doi:10.1074/jbc.RA120.014888
79. Guo Q, Zhu Q, Miao T, et al. LRP1-upregulated nanoparticles for efficiently conquering the blood-brain barrier and targetedly suppressing multifocal and infiltrative brain metastases. J Controll Rel. 2019;303:117–129. doi:10.1016/j.jconrel.2019.04.031
80. Dai T, He W, Tu S, et al. Black TiO(2) nanoprobe-mediated mild phototherapy reduces intracellular lipid levels in atherosclerotic foam cells via cholesterol regulation pathways instead of apoptosis. Bioact Mater. 2022;17:18–28. doi:10.1016/j.bioactmat.2022.01.013
81. Lee SY, Ko SH, Shim JS, et al. Tumor targeting and lipid rafts disrupting hyaluronic acid-cyclodextrin-based nanoassembled structure for cancer therapy. ACS Appl Mater Interfaces. 2018;10(43):36628–36640. doi:10.1021/acsami.8b08243
82. Varan G, Akkin S, Demirturk N, et al. Erlotinib entrapped in cholesterol-depleting cyclodextrin nanoparticles shows improved antitumoral efficacy in 3D spheroid tumors of the lung and the liver. J Drug Target. 2021;29(4):439–453. doi:10.1080/1061186X.2020.1853743
83. Lee SSY, Li JJ, Tai JN, et al. Avasimibe encapsulated in human serum albumin blocks cholesterol esterification for selective cancer treatment. Acs Nano. 2015;9(3):2420–2432. doi:10.1021/nn504025a
84. Li M, Yang YT, Wei JJ, et al. Enhanced chemo-immunotherapy against melanoma by inhibition of cholesterol esterification in CD8(+) T cells. Nanomed Nanotechnol Biol Med. 2018;14(8):2541–2550. doi:10.1016/j.nano.2018.08.008
85. Xiao X, Liu Y, Guo M, et al. pH-triggered sustained release of arsenic trioxide by polyacrylic acid capped mesoporous silica nanoparticles for solid tumor treatment in vitro and in vivo. J Biomat Applicat. 2016;31(1):23–35. doi:10.1177/0885328216637211
86. Guo JL, Du XM, Huang JS, et al. Robust dual enzyme cascade-catalytic cholesterol depletion for reverse tumor multidrug resistance. Adv Healthcare Mater. 2022;11(19). doi:10.1002/adhm.202200859
87. Du B, Zheng M, Ma HZ, et al. Nanozyme-natural enzymes cascade catalyze cholesterol consumption and reverse cancer multidrug resistance. J Nanobiotechnol. 2022;20(1):209–223. doi:10.1186/s12951-022-01406-9
88. Badr-Eldin SM, Alhakamy NA, Fahmy UA, et al. Cytotoxic and pro-apoptotic effects of a sub-toxic concentration of fluvastatin on OVCAR3 ovarian cancer cells after its optimized formulation to melittin nano-conjugates. Front Pharmacol. 2021;11:642171–642182. doi:10.3389/fphar.2020.642171
89. Dehghankelishadi P, Maritz MF, Dmochowska N, et al. Formulation of simvastatin within high density lipoprotein enables potent tumour radiosensitisation. J Control Release. 2022;346:98–109. doi:10.1016/j.jconrel.2022.04.017
90. Alfaleh MA, Fahmy O, Al-Rabia MW, et al. Hybrid nanoparticulate system of Fluvastatin loaded phospholipid, alpha lipoic acid and melittin for the management of colon cancer. Sci Rep. 2022;12(1):19446–19461. doi:10.1038/s41598-022-24151-3
91. Xi Y, Yani Z, Jing M, et al. Mechanisms of induction of tumors by cholesterol and potential therapeutic prospects. Biomed Pharmacother. 2021;144:112277–112285. doi:10.1016/j.biopha.2021.112277
92. Dewidar SA, Hamdy O, Eltantawy A, et al. Effect of concomitant use of pitavastatin with neoadjuvant chemotherapy protocols in breast cancer patients: a randomized controlled clinical trial. Saudi Pharmac J. 2022;30(10):1486–1496. doi:10.1016/j.jsps.2022.07.011
93. Sharom FJ. Complex interplay between the P-glycoprotein multidrug efflux pump and the membrane: its role in modulating protein function. Front Oncol. 2014;4:41–59. doi:10.3389/fonc.2014.00041
94. He H, Hong K, Liu L, et al. Artificial high-density lipoprotein-mimicking nanotherapeutics for the treatment of cardiovascular diseases. Wiley Interdiscipl Rev Nanomed Nanobiotechnol. 2021;13(6):e1737–e1757. doi:10.1002/wnan.1737
95. Luthi AJ, Lyssenko NN, Quach D, et al. Robust passive and active efflux of cellular cholesterol to a designer functional mimic of high density lipoprotein. J Lipid Res. 2015;56(5):972–985. doi:10.1194/jlr.M054635
96. Luthi AJ, Zhang H, Kim D, et al. Tailoring of biomimetic high-density lipoprotein nanostructures changes cholesterol binding and efflux. Acs Nano. 2012;6(1):276–285. doi:10.1021/nn2035457
97. Plebanek MP, Mutharasan RK, Volpert O, et al. Nanoparticle targeting and cholesterol flux through scavenger receptor type B-1 inhibits cellular exosome uptake. Sci Rep. 2015;5:15724–15737. doi:10.1038/srep15724
98. Thaxton CS, Daniel WL, Giljohann DA, et al. Templated spherical high density lipoprotein nanoparticles. J Am Chem Soc. 2009;131(4):1384–1385. doi:10.1021/ja808856z
99. White PT, Subramanian C, Motiwala HF, et al. Natural withanolides in the treatment of chronic diseases. Adv Exp Med Biol. 2016;928:329–373. doi:10.1007/978-3-319-41334-1_14
100. Esau L, Sagar S, Bangarusamy D, et al. Identification of CETP as a molecular target for estrogen positive breast cancer cell death by cholesterol depleting agents. Genes Cancer. 2016;7(9–10):309–322. doi:10.18632/genesandcancer.122
101. Molnar E, Swamy M, Holzer M, et al. Cholesterol and sphingomyelin drive ligand-independent T-cell antigen receptor nanoclustering. J Biol Chem. 2012;287(51):42664–42674. doi:10.1074/jbc.M112.386045
102. Liu X, Zhao Z, Sun X, et al. Blocking cholesterol metabolism with tumor-penetrable nanovesicles to improve photodynamic cancer immunotherapy. Small Methods. 2022;7:e2200898. doi:10.1002/smtd.202200898
103. Sharma D, Barhwal KK, Biswal SN, et al. Hypoxia-mediated alteration in cholesterol oxidation and raft dynamics regulates BDNF signalling and neurodegeneration in hippocampus. J Neuroch. 2019;148(2):238–251. doi:10.1111/jnc.14609
104. Mahammad S, Parmryd I. Cholesterol depletion using methyl-beta-cyclodextrin. Methods Mol Biol. 2015;1232:91–102. doi:10.1007/978-1-4939-1752-5_8
105. Yamaguchi R, Perkins G, Hirota K. Targeting cholesterol with beta-cyclodextrin sensitizes cancer cells for apoptosis. FEBS Lett. 2015;589(24 Pt B):4097–4105. doi:10.1016/j.febslet.2015.11.009
106. Li YC, Park MJ, Ye SK, et al. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am J Pathol. 2006;168(4):1107–1118. doi:10.2353/ajpath.2006.050959
107. Mohammad N, Malvi P, Meena AS, et al. Cholesterol depletion by methyl-beta-cyclodextrin augments tamoxifen induced cell death by enhancing its uptake in melanoma. Mol Cancer. 2014;13:204–216. doi:10.1186/1476-4598-13-204
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.