")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 17
Revealing the Impact of Gut Microbiota on Acne Through Mendelian Randomization Analysis
Authors Ji X , Wu S, Zhao D , Bai Q, Wang Y , Gong K, Zheng H, Zhu M
Received 5 December 2023
Accepted for publication 18 January 2024
Published 8 February 2024 Volume 2024:17 Pages 383—393
DOI https://doi.org/10.2147/CCID.S451104
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Xiaotian Ji,1 Shuhui Wu,1 Dan Zhao,1 Qi Bai,1 Yun Wang,1 Ke Gong,2 Huie Zheng,1 Mingfang Zhu1
1Department of Dermatology, the Second Affiliated Hospital of Hunan University of Chinese Medicine, Changsha, People’s Republic of China; 2Department of Traditional Chinese Medicine, Cangzhou Central Hospital, Cangzhou, People’s Republic of China
Correspondence: Mingfang Zhu, Department of Dermatology, the Second Affiliated Hospital of Hunan University of Chinese Medicine, 233 Cai E North Road, Kaifu District, Changsha, People’s Republic of China, Tel +86-13907317358, Fax +86-731-84917727, Email [email protected]
Background: The association between acne and gut microbiota has garnered considerable attention; nevertheless, given the substantial diversity within gut microbiota, the precise cause-and-effect relationship linking specific microbial species to acne remains elusive. To address this gap in knowledge, our study utilized Mendelian randomization analysis to elucidate a potential causal link between gut microbiota composition and acne development while also investigating underlying mechanisms involving microbial factors associated with metabolic disorders.
Materials and Methods: The independent single nucleotide polymorphisms (SNPs) closely associated with 196 gut microbiota samples (N=18340) were selected as variable tools. The relationship between gut microbiota and acne (N=212438) was analyzed using the Twosample package in R4.3.1, employing various methods including inverse variance weighting (IVW), weighted median, MR-Egger, Simple-mode, and Weighted-mode. To ensure the stability of the estimates, a series of sensitivity analyses were conducted, such as Cochran’s Q-test, MR-Egger intercept analysis, leave-one-out analysis, and funnel plots. Additionally, the impact of each instrumental variable was calculated.
Results: In the Mendelian randomization analysis, we identified twelve microbial taxa potentially associated with acne: family.Bacteroidaceae, family.Clostridiaceae1, genus.Allisonella, genus.Bacteroides, genus.Butyricimonas, genus.Clostridiumsensustricto1, and genus.Coprococcus3. These seven bacterial groups were found to be potential risk factors for acne. Conversely, family.Lactobacillaceae and genus.Ruminococcustorquesgroup along with genus.CandidatusSoleaferrea, genus.Fusicatenibacter, family.Lactobacillaceae, and genus.Lactobacillus exhibited a protective effect against acne. Furthermore, our investigation revealed that some of these microbial taxa have been implicated in metabolic diseases through previous studies. Importantly though, no causal relationship was observed in the reverse Mendelian randomization analysis.
Keywords: acne, gut microbiota, Mendelian randomization, metabolic disease
Introduction
The pathogenesis of acne involves a multifactorial interplay, resulting in chronic inflammation within the sebaceous glandular units located in hair follicles.1 This condition is estimated to affect 9.4% of the global population and ranks as the eighth most prevalent disease worldwide,2 imposing a significant burden on the global economy. It is prone to scarring and hyperpigmentation after healing, affecting 85% of young individuals and adolescents, with its social and psychological impact surpassing its physical consequences.3–5 A questionnaire-based study revealed that 97.8% of patients postulated potential causal or exacerbating factors for acne, encompassing sleep deprivation, tobacco use, alcohol consumption, and even concurrent infections; 95% of patients held the belief that certain dietary choices and beverages contribute to the deterioration of acne; furthermore, 85.5% of participants in this investigation associated it with the consumption of fatty/fried foods,4 indicating a widespread recognition among the general public regarding the relationship between diet and acne.6 In addition to its contribution to acne development, poor diet also plays a significant role in the pathogenesis of metabolic disorders. Our research group acknowledges the interplay between acne and metabolic disorders, recognizing that gut microbiota underlies the extensive impact of diet on human health and disease.7,8 Accordingly, it is imperative to ascertain the causal relationship between gut microbiota and acne, elucidate the underlying mechanisms by which gut microbiota influences the pathogenesis of acne, investigate potential associations between acne and metabolic disorders, and develop targeted strategies for prevention and treatment of acne.
The gut microbiota, acquired at birth, constitutes an inherent component of the human body and coevolves with the host’s development.9 To a considerable extent, gut microbiota is regarded as the fundamental organ of the human body,10 playing an indispensable role in human existence. The gut microbiota constantly maintains a dynamic equilibrium and actively participates in metabolic and immune processes.11,12 However, dysregulation of the gut microbiota can compromise the integrity of the intestinal barrier, resulting in translocation of microbial communities, systemic inflammation, and metabolic disorders.13 With the progressive advancement of scholarly research on gut microbiota and dermatological conditions, it has been substantiated that disruptions in gut microbiota impact skin health,14 while conversely, the state of the skin also influences gut microbiota, thereby establishing a reciprocal relationship between them.15,16 Polkowska-Pruszyska’s team explores the correlation between alterations in gut microbiota and immune responses that lead to various skin conditions, including acne, atopic dermatitis (AD), allergies, among others.17 Moreover, compelling evidence suggests that emotions exert an influence on acne through their impact on gut microbiota, thereby implicating a potential link between gut microbiota and the development of acne.18 The evidence presented herein elucidates a robust correlation between gut microbiota and acne, thereby suggesting that dysbiosis of gut microbiota constitutes one of the etiological factors contributing to the development of acne. To comprehensively investigate the specific causal effects linking gut microbiota and acne, including delineating the precise impact of individual microbial species on acne pathogenesis, an extensive array of fundamental experimental and observational studies is still warranted. However, it should be noted that such endeavors are associated with substantial costs and inherent challenges in controlling for confounding factors.19 According to Mendel’s law of segregation and independent assortment, genetic variations are randomly allocated to gametes, and individual genes are determined at birth and remain unchanged by acquired habits, diet, living environment, etc. In the context of this study, Single Nucleotide Polymorphisms (SNPs) are employed as an Instrument of Variation (IV) in a Mendelian randomization (MR) design that mimics the approach used in Randomized Controlled Trials (RCTs). This methodology effectively mitigates confounding factors and enhances the credibility of observed causal effects. Consequently, MR studies offer a feasible means for analyzing the causal relationship between gut microbiota and acne.
Materials and Methods
Overview of Research
In this experiment, each of the 196 gut microbiota samples was considered as an independent exposure factor, and two-sample Mendelian randomization was conducted to assess its association with acne as an outcome. To meet the requirements of MR studies, three key assumptions need to be satisfied: 1) strong correlation between instrumental variables and exposure factors; 2) no correlation between instrumental variables and confounders; and 3) absence of direct correlation between instrumental variables and outcomes, with their effect on the outcome being solely mediated through exposure.
Data Sources
Acne data were obtained from the FinnGen database, which was collected in 2021 and comprised a total of 212,438 participants (N=212438). Gut microbiota data were acquired from MiBioGen, an international consortium that collects 24S rRNA gene sequencing profiles and genotyping information from 18,340 participants across multiple cohorts in the United States, Canada, Israel, South Korea, Germany, Denmark, the Netherlands, Belgium, Sweden, Finland and the United Kingdom.20
Selection of Instrumental Variables
For the initial screening of gut microbiota, a total of 211 microbiota classifications were utilized. After excluding 15 unknown classifications, the remaining 196 classifications were considered for the experiment, comprising 9 phyla, 16 orders, 20 families, and 119 genera. The details of the gut microbiota included in the study are presented in Table S1. The SNPs of these bacterial groups (n=196) were screened based on the following criteria: 1) Initially aiming for p<5×10-8 yielded an insufficient number of SNPs; thus this criterion was abandoned in favor of using p<1×10-5 as the screening threshold.21 2) The LD criterion for SNPs that met the criteria mentioned in 1) was set as r2=0.001 and kb=10,000, ensuring the independence of the identified SNPs. 3) The F statistic was calculated for each SNP, and strong instrumental variables with F>10 were selected to mitigate weak instrumental bias.22
Mendelian Randomization Studies and Sensitivity Analysis
The inverse variance weighting (IVW) method was primarily employed in this study to analyze the causal effects of gut microbiota on acne. To enhance the stability and reliability of the experimental findings, four additional analytical methods were also utilized, including the weighted median method, MR-Egger method, Simple mode method, and Weighted mode method. The criterion for determining causal effects among these five results was based on selecting the inverse variance weighting (IVW) method as it is considered the most credible.
The experiment employed Cochran’s Q statistic to quantify and assess potential heterogeneity, as well as the MR-Egger intercept test to estimate horizontal multivariate validity. To ensure result reliability, the leave-one-out method was additionally utilized to individually eliminate each SNP and evaluate its impact on overall results and heterogeneity.
Meanwhile, to mitigate errors arising from confounding factors in the experiment, we employed PhenoScanner (http://www.phenoscanner.medschl.cam.ac.uk) to query the experimentally selected SNPs as instrumental variables, ensuring that all SNPs adhere to the three fundamental assumptions of Mendelian randomization study 2 and 3.
Statistical Analysis
Considering the inclusion of n species groups at various taxonomic levels, including phylum, class, family, order, species and genus, we applied Bonferroni correction to adjust the significance thresholds with a formula of 0.05/n.23 The resulting p-values for phylum, class, family, order, species and genus were 5.56 × 10-3, 3.13 × 10-3, 2.5 × 10-3, 1.56 × 10-3, and 4.20 × 10-4 respectively.In the experimental results, we considered p-values that remained significant after applying the Bonferroni correction as statistically significant. Additionally, those with p-values less than 0.05 but did not meet the significance threshold after the Bonferroni correction were also deemed to be of statistical importance. All experiments were primarily conducted using R software (version 4.3.1) and analyzed with the Two-Sample-MR package (version 0.5.7).
Results
Description of Instrumental Variables
The screening process involved genome-wide significance threshold testing (p<1×10-5), linkage disequilibrium (LD) analysis, coordination and harmonization, MR-PRESSO test, F-value calculation, and further screening. All retained SNPs had F-statistic values greater than 10 to ensure sufficient correlation with the corresponding microbiota. Detailed information on the retained SNPs’ correlations and statistics can be found in Table S2.
Causal Impact of Gut Microbiota on Acne Vulgaris
The MR analysis revealed that family.Bacteroidaceae (95% CI: 1.03–3.70, OR=1.96, P=0.0391), family.Clostridiaceae1 (95% CI: 1.06–2.45, OR=1.61, P=0.0258), genus. Allisonella (95% CI: 1.06–1.66, OR=1.33, P=0.0122), genus.Bacteroides (95% CI: 1.03–3.70, OR=1.96, P=0.0391), genus.Butyricimonas (95% CI: 1.03–2.13, OR=1.48, P=0.0321), genus.Clostridiumsensustricto1 (95% CI: 1.03–2.51, OR=1.61, P=0.0374), genus.Coprococcus3 (95% CI: 1.44–3.83, OR=2.35 (P=0.0006) above 7 microbiota were risk factors for acne. family.Lactobacillaceae (95% CI: 0.51–0.91, OR=0.68, P=0.0094), genus.Ruminococcustorquesgroup (95% CI: 0.37–0.93 OR=0.59, P=0.0224), genus.CandidatusSoleaferrea (95% CI: 0.53–0.95, OR=0.71, P=0.0196), genus.Fusicatenibacter (95% CI: 0.40–0.86, OR=0.59, P= 0.0065), genus.Lactobacillus (95%: 0.55–0.94, OR=0.72, P=0.150). The remaining five species exhibited protective effects against acne.
The details are depicted in Figure 1, while the outcomes of each test are presented in Table S3.
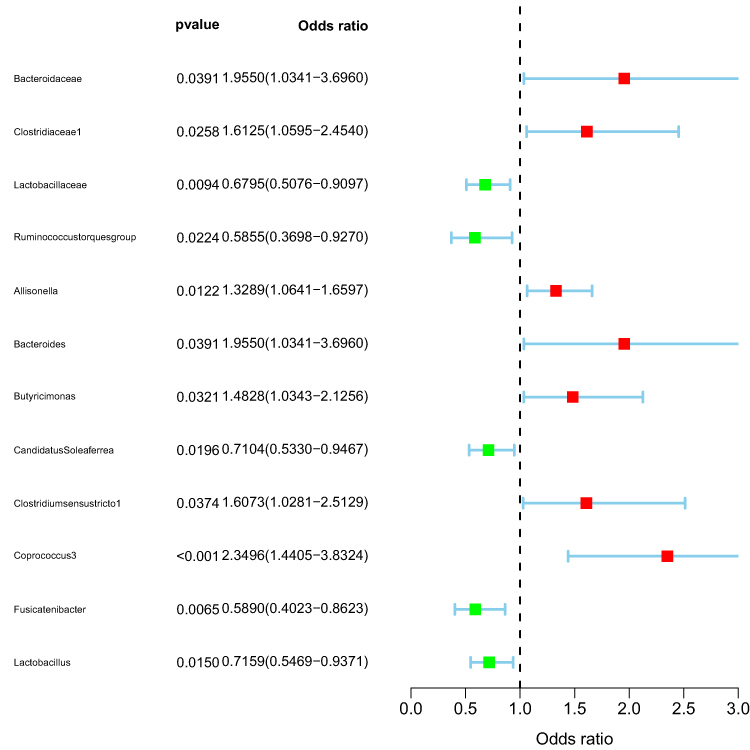 |
Figure 1 Demonstration of Cause and Effect. |
The MR-Egger method exhibited contrasting results compared to other methods in assessing the causal effect within the family Bacteroidaceae, genus. Ruminococcustorquesgroup, genus.Bacteroides, and genus.Fusicatenibacter. Conversely, the Simple-mode method demonstrated opposing outcomes when calculating the causal effect within the family.Lactobacillaceae and genus Lactobacillus. However, for the remaining five calculation methods pertaining to bacterial groups, the direction of causal effect was largely consistent.
Relevant details can be found in Figure 2
 |
Figure 2 Visualization of Results. |
Sensitivity Analysis
The detailed results of the leave-one-out test are presented in Figure 3.
 |
Figure 3 Sensitivity Analysis. |
Table 1 presents comprehensive data from all gut microbiota assays, including the results of rigorous heterogeneity and horizontal pleiotropy tests
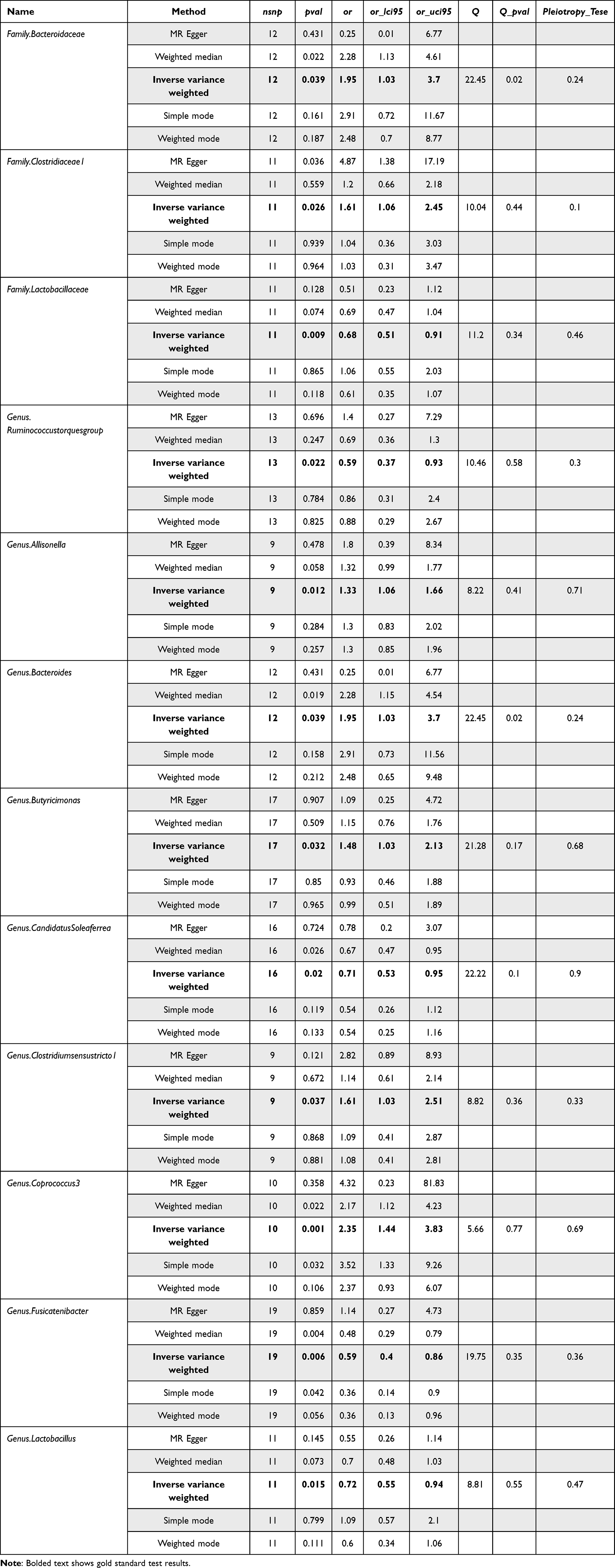 |
Table 1 Gut Microbiota Test Results |
Despite none of the p-values of the colonies tested by the IVW method reaching the Bonferroni-corrected threshold, statistical significance was still achieved due to the extremely stringent criteria. Only two out of 12 screened colonies exhibited heterogeneity, namely family.Bacteroidaceae and genus.Bacteroides, while others showed no horizontal pleiotropy and heterogeneity. Detailed data are presented in Table S4.
The analysis identified a total of 12 gut microbiota with causal effects, out of which seven exhibited risk effects and five demonstrated protective effects. Notably, the experimental results also confirmed the presence of Lactobacillus, which was found to have a mitigating effect on acne. Intriguingly, a literature review of the experimentally obtained microbiota revealed that those exhibiting a risk effect on acne were also associated with an increased risk for certain metabolic diseases. Furthermore, some consistency was observed among the microbiota displaying protective effects.
Discussion
In a previous study, which indicated a robust correlation between gut microbiota and acne development,24 we identified 12 specific microbiota that exerted a causal influence on acne through MR analysis utilizing summarized data on gut microbiota from the MiBioGen consortium and acne data from the FinnGen consortium. Among them, the bacterium exhibited a risk factor OR>1 for acne at both the family and genus levels. The genus Coprococcus3 had the highest OR, indicating that this group had the strongest impact on acne risk. Lactobacillus demonstrated a protective effect against acne at both family and genus levels, with its strongest protective effect against acne observed in the Ruminococcustorquesgroup.
The gut microbiota can be regarded as a distinct endocrine organ within the human body,25 and its composition, functional endocrine interactions, and association with obesity, cardiovascular disease, metabolic syndrome, and stress-related disorders,26 suggest an equally robust connection between acne and these diseases.27
Our study revealed that Bacteroidaceae serves as a predisposing factor for acne, with the genus comprising 78 species and 5 subspecies. Among them, B. fragile (Bacteroidaceae.fragilis) stands out as the representative species, known to induce skin and soft tissue infections.28 Furthermore, we observed that an increased abundance of B. fragile leads to a reduction in levels of bile acid glycodeoxycholic acid (GUDCA) and taurine deoxycholic acid (TUDCA), both of which act as antagonists to FXR.29 In contrast to the hepatic effects of FXR, it has been demonstrated that modulation of intestinal FXR signaling improves metabolic disorders.30–32 It is postulated that the heightened risk of acne due to an increased abundance of Bacteroidetes anthropophilus weakens the inhibitory effect on intestinal FXR, thereby significantly reducing metabolic homeostasis protection. This may be one of the underlying reasons for the comorbidity between acne and metabolic disorders.
Clostridiaceae are found to be more abundant in both inflammatory bowel disease and arthritis,33 suggesting a potential pro-inflammatory role of Clostridia. Moreover, a Mendelian randomization study has demonstrated that Clostridiaceae serve as a risk factor for T2DM.34 Our findings support the hypothesis that acne and T2DM share a common pathogenesis.27
Histamine (HA), a ubiquitous substance in living organisms, has emerged as a central neurotransmitter associated with obesity, diabetes, and endocrinology in recent years.35 The genus Allisonella has the ability to metabolize histamine,36 a component of the leptin signaling pathway,37,38 which plays a crucial role in regulating feeding behavior and metabolic processes within the body. Sebaceous glands exhibit an inflammatory response upon leptin stimulation, as indicated by augmented sebum production, activation of STAT-3 and nuclear factor (NF)-κB pathways, and enhanced secretion of cytokines IL-6 and IL-8. These findings suggest that the leptin signaling pathway may regulate sebum metabolism, thereby influencing acne development.39 Elevated concentrations of leptin in circulating or local tissues can give rise to metabolic syndrome-associated disorders, including obesity, dyslipidemia, hyperglycemia, and hypertension.40
Lactic acid bacteria, belonging to a genus of beneficial bacteria capable of lactose and protein breakdown, play a pivotal role in the gut microbiota.Lactic acid bacteria, belonging to the genus Lactobacillus, have been extensively studied for their potential anti-obesity and anti-metabolic disease properties. It has been reported that certain strains of Lactobacillus can produce short-chain fatty acids (SCFAs) through fermentation. These SCFAs play a regulatory role in signaling pathways associated with lipid synthesis, lipid metabolism, and energy regulation, including mammalian target of rapamycin (mTOR), AMP-activated protein kinase (AMPK), peroxisome proliferator-activated receptors (PPARs), among others.41–43 In 2016, Monfrecola et al demonstrated the pivotal role of the mTOR pathway in acne pathogenesis for the first time. Conversely, inhibition of the AMPK pathway upregulates sterol regulatory element-binding protein-1 (SREBP-1) expression and adipogenesis, thereby contributing to acne development.44 Lactobacillus acidophilus inhibits the mTOR pathway while activating the AMPK pathway, thus serving as a protective factor.
Butyricimonas, an enterobacterium known for its production of butyrate, has been identified as a potential risk factor for acne. However, it is noteworthy that butyrate exhibits anti-inflammatory properties by generating factors that mitigate inflammation.45 Our experimental results contradict this finding and currently there is no suitable evidence to explain the association between Butyricimonas and acne. There may be an undiscovered underlying mechanism linking them.
The study possesses several notable strengths. Firstly, the utilization of MR analysis establishes a causal relationship between gut microbiota and acne while effectively excluding confounding factors. Secondly, the inclusion of genetic variation in gut microbiota from the largest GWAS meta-analysis ensures instrumental strength in MR analysis. Additionally, horizontal pleiotropy was meticulously tested and excluded using MR-PRESSO and MR-Egger regression intercept terms.
However, there are certain limitations that should be acknowledged: (1) The absence of Asian-African populations participating in both the enterobacterial database and acne database used in this experiment raises uncertainty regarding the generalizability of these findings to global human populations. (2) Due to stringent experimental settings for P thresholding and removal of linkage disequilibrium, it is possible that some equally effective SNPs were inadvertently excluded. (3) Despite careful manual screening of SNPs, it is important to acknowledge that complete elimination of all potential confounding factors may not have been achieved by the authors, potentially introducing bias into the results.
Conclusion
In summary, our findings indicate a strong causal relationship between acne-associated microbiota and metabolic diseases, including diabetes and cardiovascular diseases. This high correlation underscores the need for further research to elucidate the underlying mechanisms linking acne and metabolic disorders. Additionally, our study provides valuable insights into both beneficial and risky microbiota associated with acne, offering potential guidance for clinical treatment and prevention strategies. Furthermore, we explore the possible connections between acne and metabolic diseases, investigating shared etiological factors and proposing avenues for studying comorbidities of acne. These contributions highlight the significance of this paper.
Data Sharing Statement
The dataset utilized in this study is available for download from the MiBioGen repository (https://mibiogen.gcc.rug.nl/), and FinnGen repository (https://r9.finngen.fi/).
Ethics Statement
According to Article 32 of the Measures for Ethical Review of Life Science and Medical Research Involving Human Beings adopted by the National Science and Technology Ethics Committee of the People’s Republic of China, ethical review can be exempted because the data used in this study do not cause any harm to human beings, do not involve any sensitive personal information or commercial interests, and the databases selected are open and legal.
Acknowledgments
The authors extend their sincere gratitude to the participants and investigators of the FinnGen study. Furthermore, the authors would like to express their appreciation to the MiBioGen consortium for providing access to the gut microbiota GWAS summary statistics.
Funding
This work was funded by the National Natural Science Foundation of China (82174375), Scientific Research Project of Hunan Provincial Health Commission (202202124772), Key Project of Hunan Provincial Administration of Traditional Chinese Medicine (c2022026), Hunan Provincial Natural Science Foundation of Science and Technology and Medicine Joint Fund Project (2020SK51301), Hunan Provincial Clinical Medicine Technology Innovation Guidance Project (2020SK51301) and Hunan Provincial Key Laboratory of Vascular Biology and Translational Medicine.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fox L, Csongradi C, Aucamp M, du Plessis J, Gerber M. Treatment Modalities for Acne. Molecules. 2016;21(8):1063.
2. Tan JK, Bhate K. A global perspective on the epidemiology of acne. Br J Dermatol. 2015;172 Suppl 1:3–12.
3. Thiboutot D, Gollnick H, Bettoli V, et al. New insights into the management of acne: an update from the Global Alliance to Improve Outcomes in Acne group. J Am Acad Dermatol. 2009;60(5 Suppl):S1–50.
4. Yorulmaz A, Yalcin B. Myths, Perceptions and Practices in Acne: a Study on Adolescents and Young Adults. Curr Health Sci J. 2020;46(2):111–116.
5. Dall’Oglio F, Nasca MR, Fiorentini F, Micali G. Diet and acne: review of the evidence from 2009 to 2020. Int J Dermatol. 2021;60(6):672–685.
6. Baldwin H, Tan J. Effects of Diet on Acne and Its Response to Treatment. Am J Clin Dermatol. 2021;22(1):55–65.
7. Gentile CL, Weir TL. The gut microbiota at the intersection of diet and human health. Science. 2018;362(6416):776–780.
8. Moszak M, Szulińska M, Bogdański P. You Are What You Eat-The Relationship between Diet, Microbiota, and Metabolic Disorders-A Review. Nutrients. 2020;12(4):1096.
9. Adak A, Khan MR. An insight into gut microbiota and its functionalities. Cell Mol Life Sci. 2019;76(3):473–493.
10. Ding RX, Goh WR, Wu RN, et al. Revisit gut microbiota and its impact on human health and disease. J Food Drug Anal. 2019;27(3):623–631.
11. Musso G, Gambino R, Cassader M. Obesity, diabetes, and gut microbiota: the hygiene hypothesis expanded. Diabetes Care. 2010;33(10):2277–2284.
12. Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature. 2012;489(7415):231–241.
13. Festi D, Schiumerini R, Eusebi LH, Marasco G, Taddia M, Colecchia A. Gut microbiota and metabolic syndrome. World J Gastroenterol. 2014;20(43):16079–16094.
14. Pascal M, Perez-Gordo M, Caballero T, et al. Microbiome and Allergic Diseases. Front Immunol. 2018;9:1584.
15. Salem I, Ramser A, Isham N, Ghannoum MA. The Gut Microbiome as a Major Regulator of the Gut-Skin Axis. Front Microbiol. 2018;9:1459.
16. Levkovich T, Poutahidis T, Smillie C, et al. Probiotic bacteria induce a ‘glow of health’. PLoS One. 2013;8(1):e53867.
17. Polkowska-Pruszyńska B, Gerkowicz A, Krasowska D. The gut microbiome alterations in allergic and inflammatory skin diseases - an update. J Eur Acad Dermatol Venereol. 2020;34(3):455–464.
18. Bowe W, Patel NB, Logan AC. Acne vulgaris, probiotics and the gut-brain-skin axis: from anecdote to translational medicine. Benef Microbes. 2014;5(2):185–199.
19. de la Torre Hernández JM, Edelman ER. From Nonclinical Research to Clinical Trials and Patient-registries: challenges and Opportunities in Biomedical Research. Rev Esp Cardiol. 2017;70(12):1121–1133.
20. Kurilshikov A, Medina-Gomez C, Bacigalupe R, et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet. 2021;53(2):156–165.
21. Sanna S, van Zuydam NR, Mahajan A, et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat Genet. 2019;51(4):600–605.
22. Burgess S, Thompson SG. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40(3):755–764.
23. Curtin F, Schulz P. Multiple correlations and Bonferroni’s correction. Biol Psychiatry. 1998;44(8):775–777.
24. Sánchez-Pellicer P, Navarro-Moratalla L, Núñez-Delegido E, Ruzafa-Costas B, Agüera-Santos J, Navarro-López V. Acne, Microbiome, and Probiotics: the Gut-Skin Axis. Microorganisms. 2022;10(7):1303.
25. Clarke G, Stilling RM, Kennedy PJ, Stanton C, Cryan JF, Dinan TG. Minireview: gut microbiota: the neglected endocrine organ. Mol Endocrinol. 2014;28(8):1221–1238.
26. Tremaroli V, Bäckhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489(7415):242–249.
27. Wang Y, Zhu M, Wu S, Zheng H. Acne Comorbidities. Clin Cosmet Invest Dermatol. 2022;15:2415–2420.
28. Wexler HM. Bacteroides: the good, the bad, and the nitty-gritty. Clin Microbiol Rev. 2007;20(4):593–621.
29. Sun L, Xie C, Wang G, et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat Med. 2018;24(12):1919–1929.
30. Chiang J, Ferrell JM. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am J Physiol Gastrointest Liver Physiol. 2020;318(3):G554–G573.
31. Trabelsi MS, Daoudi M, Prawitt J, et al. Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nat Commun. 2015;6:7629.
32. Li F, Jiang C, Krausz KW, et al. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat Commun. 2013;4:2384.
33. Muñiz Pedrogo DA, Chen J, Hillmann B, et al. An Increased Abundance of Clostridiaceae Characterizes Arthritis in Inflammatory Bowel Disease and Rheumatoid Arthritis: a Cross-sectional Study. Inflamm Bowel Dis. 2019;25(5):902–913.
34. Sun K, Gao Y, Wu H, Huang X. The causal relationship between gut microbiota and type 2 diabetes: a two-sample Mendelian randomized study. Front Public Health. 2023;11:1255059.
35. Watanabe T, Yanai K. Studies on functional roles of the histaminergic neuron system by using pharmacological agents, knockout mice and positron emission tomography. Tohoku J Exp Med. 2001;195(4):197–217.
36. Garner MR, Gronquist MR, Russell JB. Nutritional requirements of Allisonella histaminiformans, a ruminal bacterium that decarboxylates histidine and produces histamine. Curr Microbiol. 2004;49(4):295–299.
37. Furusawa T, Naka I, Yamauchi T, et al. The Q223R polymorphism in LEPR is associated with obesity in Pacific Islanders. Hum Genet. 2010;127(3):287–294.
38. Etemad A, Ramachandran V, Pishva SR, et al. Analysis of Gln223Agr polymorphism of Leptin Receptor Gene in type II diabetic mellitus subjects among Malaysians. Int J Mol Sci. 2013;14(9):19230–19244.
39. Törőcsik D, Kovács D, Camera E, et al. Leptin promotes a proinflammatory lipid profile and induces inflammatory pathways in human SZ95 sebocytes. Br J Dermatol. 2014;171(6):1326–1335.
40. Hong CJ, Lin CH, Yu YW, Chang SC, Wang SY, Tsai SJ. Genetic variant of the histamine-1 receptor (glu349asp) and body weight change during clozapine treatment. Psychiatr Genet. 2002;12(3):169–171.
41. Castañeda-Márquez AC, Díaz-Benítez CE, Bahena-Roman M, et al. Lactobacillus paracasei as a protective factor of obesity induced by an unhealthy diet in children. Obes Res Clin Pract. 2020;14(3):271–278.
42. Li Y, Liang X, Lyu Y, et al. Association between the gut microbiota and nonalcoholic fatty liver disease: a two-sample Mendelian randomization study. Dig Liver Dis. 2023.
43. Wang H, Cheng X, Zhang L, Xu S, Zhang Q, Lu R. A surface-layer protein from Lactobacillus acidophilus NCFM induces autophagic death in HCT116 cells requiring ROS-mediated modulation of mTOR and JNK signaling pathways. Food Funct. 2019;10(7):4102–4112.
44. Monfrecola G, Lembo S, Caiazzo G, et al. Mechanistic target of rapamycin (mTOR) expression is increased in acne patients’ skin. Exp Dermatol. 2016;25(2):153–155.
45. Salvi PS, Cowles RA. Butyrate and the Intestinal Epithelium: modulation of Proliferation and Inflammation in Homeostasis and Disease. Cells. 2021;10(7):1775.
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.