")
Back to Journals » Clinical and Experimental Gastroenterology » Volume 17
Second-Line Treatment of Pancreatic Adenocarcinoma: Shedding Light on New Opportunities and Key Talking Points from Clinical Trials
Authors Imperial R, Mosalem O , Majeed U, Tran NH, Borad MJ, Babiker H
Received 16 September 2023
Accepted for publication 11 April 2024
Published 18 April 2024 Volume 2024:17 Pages 121—134
DOI https://doi.org/10.2147/CEG.S390655
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Vipul Yagnik
Robin Imperial,1 Osama Mosalem,1 Umair Majeed,1 Nguyen H Tran,2 Mitesh J Borad,3 Hani Babiker1
1Division of Hematology and Oncology, Department of Medicine, Mayo Clinic, Jacksonville, FL, USA; 2Department of Medical Oncology, Rochester, MN, USA; 3Division of Hematology and Oncology, Department of Medicine, Mayo Clinic, Phoenix, AZ, USA
Correspondence: Hani Babiker, Division of Hematology and Oncology, Department of Medicine, Mayo Clinic, 4500 San Pablo Rd S, Jacksonville, FL, 32224, USA, Tel +904 953 2000, Fax +904-953-2315, Email [email protected]
Abstract: Despite improvements in overall cancer mortality, deaths related to pancreatic cancer continue to rise. Following first-line treatment, second-line options are significantly limited. Classically, first-line treatment consisted of either gemcitabine or 5-fluorouracil based systemic chemotherapy. Upon progression of disease or recurrence, subsequent second-line treatment is still gemcitabine or 5-fluorouracil based chemotherapy, depending on what was used in the first line and the timing of progression or recurrence. A better understanding of the molecular underpinnings of pancreatic adenocarcinoma (PDAC) has led to new treatment strategies including specifically targeting the desmoplastic stroma, cytokine signaling and actionable mutations. Furthermore, efforts are also directed to enhance the immunogenicity profile of PDAC’s well-established immunologically “cold” tumor microenvironment. More recently, the outstanding response rates of chimeric antigen receptor T (CAR-T) cells in hematologic malignancies, have led to clinical trials to evaluate the treatment modality in PDAC. In this review, we summarize recently presented clinical trials for metastatic pancreatic adenocarcinoma with novel treatment approaches in the second line and beyond.
Keywords: pancreatic adenocarcinoma, immunotherapy, CAR-T, KRAS
Introduction
Epidemiology
Pancreatic adenocarcinoma (PDAC) is the 10th most common cancer but the 3rd deadliest cancer affecting an estimated 64,050 new patients in 2023.1 Incidence rates have continued to rise by approximately 1% every year since 2000 despite significant technological advances in diagnostic evaluation and treatment strategies of cancers including next-generation sequencing, targeted therapies, and immunotherapy. The most recent cancer outcomes statistics published by the ACS in 2023 found that, although cancer death rates have been decreasing, PDAC death rates have been slowly increasing.1 For all stages combined, the 5-year survival rate is 11–12.5%.1,2 Most cases are diagnosed in late stage. Only 13% of patients have localized/early-stage disease at diagnosis and even in this population, the 5-year survival rate is 42%. In the setting of metastatic disease (mPDAC), 5-year survival rates are an abysmal 3.1%. Following first line chemotherapy with gemcitabine plus nab-paclitaxel or FOLFIRINOX derivative chemotherapy regimens, limited options exist for second-line therapy, and outcomes are far more dismal.
Tumor Biology
The hallmark of PDAC tumors is the desmoplastic stroma that surrounds the tumor cells and makes up approximately 70% of the mass itself.3 It serves multiple functions including providing a microenvironment that is active in tumorigenesis and creating a physical barrier of fibrosed tissue that limits perfusion of blood and nutrients. As such, diffusion of anti-tumor therapies is also limited. Hyaluronic acid, a large glycosaminoglycan present in the stroma, has been associated with resistance to chemotherapy in preclinical PDAC models.4 Moreover, the subsequent hypoxic microenvironment created by limited perfusion leads to inhibition of effector-T cells via increased regulatory-T cell mediated mechanisms and leads to a profound effect on immunogenicity.5 These immunologically “cold” tumors can evade the immune system and demonstrate poor response rates to immune checkpoint inhibition (ICI).
In an ongoing study presented by at the ASCO Annual Meeting in 2021, Rahma et al conducted a Phase Ib/II randomized control trial of neoadjuvant chemoradiation with or without pembrolizumab in patients with resectable or borderline resectable PDAC (NCT02305186).6 The goal of the study was to determine safety and efficacy of neoadjuvant chemotherapy combined with ICI, pembrolizumab. In previous studies, other solid tumors treated with neoadjuvant chemotherapy ± radiation have resulted in increased recruitment of tumor infiltrating lymphocytes in the tumor microenvironment.7–10 However, preliminary data from Rahma et al did not demonstrate differences in activated CD8 T cells or regulatory T cell populations in any of the thirty-seven PDAC patients who received pembrolizumab in combination with chemoradiation. This suggests that PDAC has a unique tumor microenvironment that creates an immunologically “cold” clinical phenotype.
More strategies are needed to modulate and enhance the immunogenicity of PDAC. As evidenced in a study by Balachandran et al.11 This retrospective study used molecular analysis to characterize the PDAC’s immunogenic profile in patients who did not receive neoadjuvant chemotherapy. Patients were divided into long-term survivors (LTS) with an overall survival greater than 3 years from surgery or short-term survivors (STS) with an overall survival between 3 months and 1 year. The tumor immunogenic phenotypes of these two patient populations were compared. LTS tumors exhibited an enhanced immunologically “hot” profile characterized by a 3-fold increase in density of CD8+ T cells, a 12-fold increase in cytolytic CD8+ T cells, and an increase in regulatory T cells, mature dendritic cells and macrophages compared to STS which had a higher density of CD4+ T cells. Furthermore, transcriptomic analysis found LTS tumor microenvironments to have up-regulation of markers of dendritic cells and PD-1 and TIGIT expression. In addition, neoantigen prediction algorithms found that neoantigen quality and quantity generated by the tumor combined with higher CD8+ T-cell infiltration correlated with survival. Overall, these results suggest that raising the immunogenic profile may correlate with improved outcomes.
Genomic analysis by next generation sequencing studies of PDAC have identified multiple driver mutations. The most mutated gene is KRAS (Kirsten rat sarcoma oncogene) which occurs in over 90% of sequenced PDAC and represents the major molecular driver of oncogenesis.12 KRAS mutations (KRASmut) result in the constitutive activation of the RAF/MEK/ERK and PIK3/AKT/mTOR pathways.13 In addition to promoting tumor growth, downstream effects of these pathways also contribute to the immunogenicity of PDAC. A study of KRAS-deficient PDAC xenografts in immunodeficient mice retained the ability to form tumors.14 However, when the isogenic KRAS knockout xenografts were introduced in immunocompetent mice, a strong anti-tumor response was observed.
At the molecular level, KRAS mutations have multiple immunologically relevant downstream effects. They lead to an increased production of GM-CSF which recruits immunosuppressive myeloid cells and the production and maintenance of the desmoplastic stroma. CD73, a cell surface 5’-nucleotidase, is up-regulated and participates in generating extracellular adenosine which can promote intrinsic or therapy-induced immune escape through multiple mechanisms.15 Interestingly, KRAS is also involved in MHC presentation. KRAS G12C inhibition has been shown to induce MHC class I presentation of haptenated peptide neoepitopes.16 KRAS has also been linked to up-regulation of CD47, an antiphagocytic cell marker which allows the tumor cells to evade the innate immune system.17
The most common mutated residue is the G12 codon and the most prevalent KRAS mutation is G12D (39.2%) followed by G12V (32.5%), G12R (17.1%), Q61H (4.8%), G12C (1.7%)(Figure 1).18 G12 mutations differ in outcomes. A retrospective study done at MD Anderson of 578 patients with KRAS mutated PDAC demonstrated significantly worse survival irrespective of stage IV patients with KRAS G12D and G12V compared to wild-type.19 KRAS G12C was too few in frequency to statistically assess. KRAS G12R appear to have similar overall survival to that of wild-type. KRAS G12 mutations have been shown to also differ in PD-L1 expression as determined by immunohistochemical staining.20 KRAS G12C demonstrated the highest PD-L1 positivity (23.5%) followed by G12D (19%), G12V (14.2%) and G12R (13%). Altogether, these results suggest that targeting KRAS could improve the immunogenicity of the tumor.
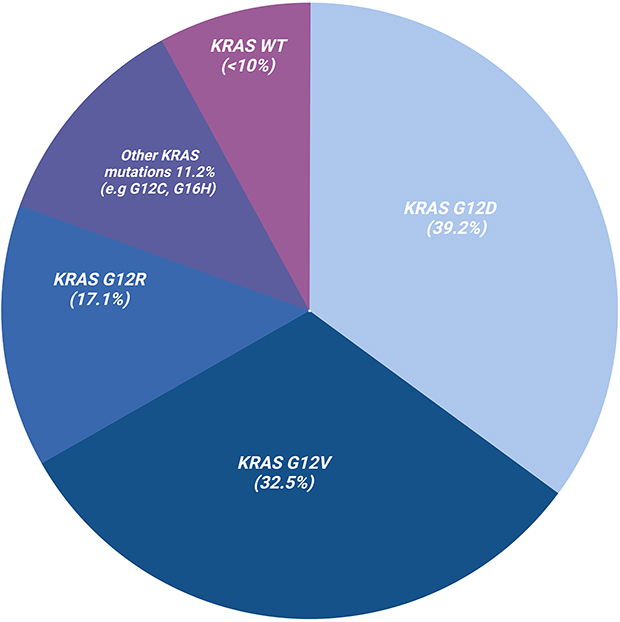 |
Figure 1 KRAS mutations in pancreatic adenocarcinoma (PDAC). Over 90% of PDAC are KRAS mutant with G12D being the most common (39.2%), followed by G12V (32.5%), G12R (17.1%), G16H (4.8%) as well as G12C (1.7%). Wild-type KRAS are less frequent and seen in < 10% of PDAC cases. |
KRAS wild-type (KRASwt) PDAC represent a small proportion of PDAC (<10%) and can be divided into 3 groups based on potential personalized therapeutics: MAPK pathway-activated (3–4% of PDAC), kinase-fusion (4% of PDAC) and microsatellite instability (1–2% of PDAC).21 In the absence of KRAS mutations, activation of MAPK is present in approximately 30% of cases. The most common alteration seen in this scenario has been BRAF mutations which is a component of the MAPK pathway downstream of KRAS. BRAF mutations are mutually exclusive of KRAS mutations. Other alterations include mutations or amplifications in genes GNAS, EGFR, ERBB2, MET, ERBB3, and FGFR2. Most kinase fusion alterations in PDAC occur with the fusion of a serine/threonine kinase domain to an oligomerization domain resulting in constitutive activation of the kinase domain. These events involve kinase domains of RET, ALK, NTRK1, NRG1, ERBB4, FGFR3, and RAF. They are mutually exclusive of each other and have not been present in patients with KRAS mutations. Microsatellite instability-high (MSI-H) is the result of defects in DNA mismatch repair genes MLH1, MSH1, MSH6, and PMS2. It is a molecular phenotype with immunotherapy implications across all cancer types.
Standard of Care
Following first line chemotherapy, current standard of care options are limited in PDAC. The most recent NCCN guidelines for second line treatment recommend the use of gemcitabine-based chemotherapy if fluoropyrimidine-based chemotherapy was used first line and vice-versa.22
Alternatively, nanoliposomal irinotecan (NAL-IRI) + 5-fluorouracil and leucovorin (5-FU/leucovorin) received FDA approval in the second line for patients who received gemcitabine-based chemotherapy in the first line.22 Delivery of irinotecan by nanoliposomal carrier resulted in a 5.6-fold higher intratumoral level of SN-38, the active metabolite of irinotecan, compared to the intravenous formulation of irinotecan alone at 72 hours.23 In a Phase II study, NAL-IRI demonstrated significant activity in metastatic PDAC patients previously treated with gemcitabine-based therapies (median overall survival (mOS) 5.2 months, 1-year overall survival (OS) 25%, NCT04005339)24 and led to the global, Phase III, NAPOLI-1 trial (NCT01494506).25 This study compared 3 arms: NAL-IRI + 5-FU/leucovorin, 5-FU/leucovorin alone and NAL-IRI alone. Four hundred seventeen patients were randomized 1:1:1 to each arm. NAL-IRI + 5-FU/leucovorin had a median OS of 6.1 months compared to 4.2 months for 5-FU/leucovorin alone (hazard ratio (HR) 0.67, P = 0.012). NAL-IRI monotherapy did not differ from 5-FU/leucovorin monotherapy arm (4.9 months vs 4.2 months, P = 0.94). This study led to the FDA approval of NAL-IRI + 5-FU in mPDAC following progression on gemcitabine-based chemotherapy in 2015.
Moreover, based on the results of the NAPOLI-3 (NCT04083235) trial, the FDA approved NALIRIFOX as a first-line option in metastatic PDAC as of February 13th, 2024.26 Data presented at the 2023 ASCO Gastrointestinal Cancers Symposium demonstrated improved mOS (11.1 months vs. 9.2 months, HR 0.834, P=0.0355) and median progression free survival (mPFS, 7.4 months vs. 5.6 months, HR 0.6944, P < 0.0001) in favor of patients treated with nanoliposomal irinotecan combined with 5-FU/leucovorin and oxaliplatin (NALIRIFOX) compared to patient treated with gemcitabine plus nab-paclitaxel.27 Sequencing gemcitabine-based, 5-FU-based, and NALIRI-based therapies will become a topic of great interest.
Recent advances and widespread availability of molecular and genomic analysis in cancer have opened the door for modern therapeutic strategies beyond the current standard chemotherapy options in cancer. Recent technologies such as organoid modeling and single-cell sequencing have led to better characterization of tumor microenvironment dynamics and responses to investigational therapeutics. Genomic sequencing has identified targetable alterations and spurred drug development. Harnessing one’s own immune system with immunotherapies such as immune checkpoint inhibitors has been readily adapted in the standard of care treatments for other malignancies. Furthermore, personalized vaccines are in development based on the genomic characteristics of an individual’s malignancy. This is a “Golden Age” of therapeutic development for which PDAC has lagged behind. In this review, we will highlight recent clinical trials and novel therapeutics for second-line consideration in PDAC.
Approaches to Second-Line Therapeutics
Chemotherapeutic Agents
Several trials have been conducted to potentiate the effects of existing chemotherapeutic options. In the phase II SEQUENCE trial investigators sought to determine the efficacy and feasibility of alternating first line treatments each cycle rather than using each respective first line, gemcitabine with nab-paclitaxel vs modified FOLFOX agents in succession on progression of disease (NCT02504333).28 One hundred fifty-seven patients were randomized 1:1 to nab-paclitaxel (125 mg/m2) plus gemcitabine (1000 mg/m2) on days 1, 8, 15, followed by modified FOLFOX-6 (5-fluorouracil bolus (400 mg/m2), and 5-fluorouracil 48-hour continuous infusion (2400 mg/m2), L-leucovorin (200 mg/m2) or racemic leucovorin (400 mg/m2), and oxaliplatin (85 mg/m2) on day 29 of a 6 week cycle or nab-paclitaxel (125 mg/m2) plus gemcitabine (1000 mg/m2) on days 1, 8, and 15 of a 4 week cycle.
The SEQUENCE trial met its primary endpoint of overall survival. 12-month overall survival rates were higher in the combination arm (55.3%) compared to the nab-paclitaxel/gemcitabine arm (35.4%, P = 0.016). mOS was 13.2 months compared to 9.7 months (HR 0.676, P=0.023). However, from a safety standpoint, the combination arm had significantly more grade 3 or higher cytopenias (neutropenia 46% vs 24%, P = 0.004; thrombocytopenia 24% vs 8%, P = 0.007). Further, two patients in the combination arm died from overwhelming infection. Logistically, the combination arm resulted in prolonged exposure to chemotherapy. Those in the combination arm stayed on treatment on average 8.3 months compared to 4.0 months in the nab-paclitaxel/gemcitabine arm. There were more dose delays, dose reductions, and transient interruptions in the experimental arm. However, EORTC QLC-C30 quality of life metrics did not identify any significant differences in quality of life between the two arms
To help potentiate the effects of gemcitabine, a metabolic enzyme inhibitor GP-2250 is currently undergoing a phase 1/2 trial (NCT03854110).29 GP-2250 has been shown in preclinical models to inhibit glyceraldehyde-3-phosphate dehydrogenase, thus limiting aerobic glycolysis, and selectively inducing oxidative stress, mitochondrial dysfunction, and apoptosis.30 When combined with gemcitabine, the drugs act synergistically in a dose-dependent manner. The open-label phase 1/2 trial of GP-2250 with gemcitabine is underway in patients with advanced unresectable or mPDAC who have progressed on prior FOLFIRINOX. The last updated status as of this writing indicates that the trial is still recruiting.
Constitutive KRAS activation is associated with altered metabolic pathways, resulting in increased dependence on metabolites, including glutamine and asparagine.31 Conceivably, decreasing levels of glutamine and asparagine may affect the downstream effects of KRAS activation. Asparaginase is an enzyme involved in the breakdown of asparagine and has been used in the treatment of acute lymphoblastic leukemia.32 While its effectiveness in solid tumors has not yet been established, authors of a recent phase II study evaluated asparaginase in PDAC.33 Given its particularly narrow therapeutic index, asparaginase was encapsulated in erythrocytes (eryaspase) through a proprietary process, resulting in a drug delivery system with lower toxicities. This study randomized one hundred forty-one patients 2:1 to eryaspase with gemcitabine or 5-FU based chemotherapy vs gemcitabine or 5-FU chemotherapy alone. Patients in the eryaspase with chemotherapy arm had a mOS of 6.0 months vs 4.4 months with chemotherapy alone (HR 0.60, P = 0.008). mPFS was 2.0 months vs 1.6 months (HR 0.56, P = 0.005). The results of this study led to the Phase III Trybeca-1 clinical trial.
TRYbeCA-1 was a randomized, open-label phase III clinical trial of eryaspase with gemcitabine/nab-paclitaxel or 5-FU/leucovorin/irinotecan chemotherapy compared to chemotherapy alone in patients with stage III or IV PDAC who had progressed on first line chemotherapy (NCT03665441).34 Five hundred twelve patients were randomized 1:1 to receive eryaspase with chemotherapy vs chemotherapy alone. This study did not meet its primary endpoint of overall survival. The mOS of the eryaspase with chemotherapy arm was 7.5 months compared to 6.7 months for chemotherapy alone (HR 0.92, P=0.375). However, there did appear to be some benefit in subgroup analysis. Eryaspase with 5-FU/leucovorin/irinotecan had a mOS of 8.0 months compared to 5.7 months for 5-FU/leucovorin/irinotecan alone (HR 0.81).
Targeted Therapies
Targeted therapies against driver mutations can be thought of as KRAS mutated (KRASmut) or KRAS wild-type (KRASwt) alterations. KRASmut represent the major molecular driver of PDAC (>90% of all sequenced PDAC).18,35 As of right now, there are no FDA approved treatments for KRASmut PDAC. However, MRTX-1133, a potent, noncovalent KRAS G12D-specific inhibitor is currently in Phase I clinical trials that has promising preclinical data. The drug was shown to be efficacious in KRAS G12D mutant xenograft mouse models with −62% and −73% tumor regressions observed at doses of 10 and 30 mg/kg, respectively.36
Two drugs targeting KRAS G12C, sotorasib and adagrasib, recently made it to phase II and III clinical trials in solid malignancies and have received FDA approval as a second-line treatment in non small cell lung cancer. Both are small molecules that form an irreversible covalent bond specifically to the KRAS G12C mutant allele.
Sotorasib was studied in CodeBreak-100 (NCT03600883).37 It was a phase I/II clinical trial evaluating the safety, and efficacy of sotorasib in patients with solid malignancies harboring the KRAS G12C mutation who progressed on at least one line of treatment. Thirty-eight PDAC patients were enrolled in the study. Eight patients (21%) had centrally confirmed objective response (complete response or partial response). mOS was 6.9 months and mPFS was 4.0 months. Six patients (15.7%) had grade 3 or higher adverse events. There were no treatment-related fatalities.
Adagrasib was studied in KRYSTAL-1 (NCT03785249).38 KRYSTAL-1 was also a phase I/II clinical trial designed to evaluate the safety and efficacy of the drug in solid malignancies with KRAS G12C mutations. Thirty patients were enrolled in the study, ten of which were PDAC. Data updated from October 1, 2022, revealed that five out of the ten patients with PDAC (50%) had an objective response (OR). All were partial responses. Interestingly, the remaining five patients who did not have a partial responsedid have stable disease, which resulted in a disease control rate (DCR) of 100% as defined by (CR + PR + SD). The median duration of response (mDOR) was 7.0 months, median progression-free survival (mPFS) was 6.6 months and median overall survival (mOS) was 8 months. Although response rates are not as robust as was seen in non-small cell lung cancer, in the context of previously treated PDAC, the response rates are significant and encouraging.
For patients with KRASwt (<10% of PDAC), there may be a role for targeted drugs in patients as rare targetable alterations can occur in absence of KRAS mutations. Neuregulin 1 (NRG1) gene fusions have been identified in <1% of PDAC.39,40 NRG1 is known to bind the ERBB3 receptor resulting in heterodimerization with ERBB2 to activate downstream signaling pathways including RAS, MAPK and PI3K.41 Gene fusions with NRG1 lead to an increased gene expression and may represent a new actionable mutation. As proof of concept, a phase I clinical trial of an anti-ERBB3 monoclonal antibody used to treat patients with ERBB3 overexpression in solid tumors identified an exceptional responder who harbored an NRG1 gene fusion (NCT01966445).42 Furthermore, another proof-of-concept study used a bispecific antibody, zenocutuzumab (MCLA-128), to treat two patients PDAC patients with wild-type KRAS and NRG1 fusions.43 Zenocutuzumab interacts with HER2 and blocks the binding of NRG1 to HER3 thus preventing the heterodimerization of HER2 and HER3 and downstream signaling. The first patient had a 54% decrease in tumor diameter and 81% decrease in CA 19–9. The second patient had a 25% decrease in tumor diameter and 97% decrease in CA 19–9. Both remained on treatment for 7-plus months. A third patient with NSCLC had a partial response. This series of patients was presented at the 2019 AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics in Boston, MA on October 26–30.
Results from the Phase I–II eNRGy trial (NCT02912949) led to the FDA giving breakthrough designation to zenocutuzumab in patients with advanced unresectable or metastatic NRG1 fusion-positive PDAC that progressed following prior systemic therapy or for which there are no suitable alternative. This open-label, multi-center trial assessed response to zenocutuzumab in NRG1-positive cancers including PDAC, NSCLC and other solid tumors.44 As of January 12, 2022, ninety-nine patients with NRG1+ cancer were enrolled including eighteen PDAC. Seventy-three patients received at least one dose of zenocutuzumab and had at least 6 months of follow up. Overall response rate was 34% (90% CI: 25–44%). Specifically, in PDAC, seven out of eighteen patients had responses (39%). The median duration of response for the seventy-three patients was 9.1 months. The treatment was well tolerated with grade 3 or higher treatment related adverse events present in less than 5% of patients. As of June 1, 2023, over one hundred seventy-five patients have been treated with zenocutuzumab monotherapy.
Other driver mutations with FDA approved targeted drugs in metastatic solid tumors include RET fusions45 (<1% of PDAC), ALK rearrangements46 (<1% of PDAC), FGFR2 fusions47 (5% of PDAC) and NTRK fusions48 (5% of PDAC) (Figure 2).
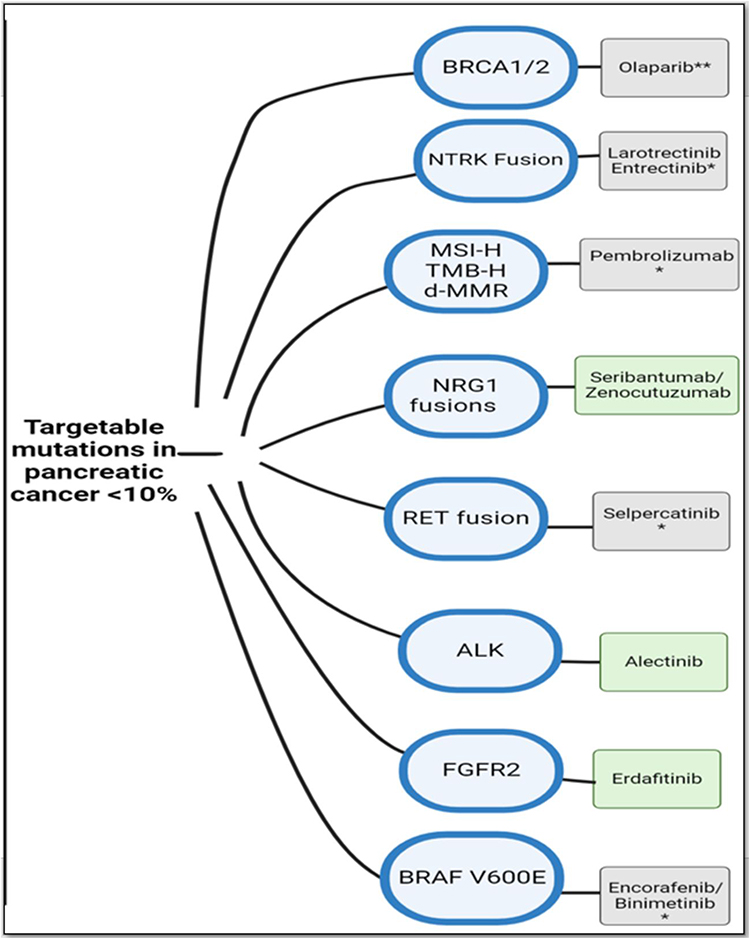 |
Figure 2 Potential targetable mutations in PDAC along with examples of drugs in each category. Targetable mutations are seldom found in PDAC, and likely to present in KRAS wild-type status (<10%). *Approved tissue-agnostic therapies are denoted in gray; those in the green box are still not FDA-approved. **Olaparib is FDA approved as a maintenance regimen in those with germline BRCA mutations which have not progressed on at least 16 weeks of first line platinum-based chemotherapy. |
Outside of driver mutations, a novel approach to targeted drug therapy is specifically targeting metabolic pathways. PDAC cells exhibit complex reprogramming of their metabolic pathways including glucose, amino acid, and lipid metabolism.49 Pyruvate dehydrogenase kinase (PDK) is a mitochondrial enzyme involved in the conversion of cytosolic pyruvate to mitochondrial acetyl-CoA. Inhibition of this enzyme with an orphan drug, Dichloroacetate (DCA), FDA approved for congenital lactic acidosis shifts cancer metabolism from glycolysis to mitochondrial oxidation as well as reverses the suppression of mitochondria-dependent apoptosis.50 It has shown promising preclinical results in PDAC cell-lines and is currently being studied in glioblastoma in phase II clinical trial (NCT05120284).50–52 Another PDK inhibitor, 3-amino-1,2,4-triazine, has also shown promising preclinical results in PDAC cell lines; however, it has not been evaluated in clinical trials yet.53
Targeting the desmoplastic stroma as an adjunct treatment to systemic chemotherapy or immunotherapy is another approach. Focal Adhesion Kinase (FAK) is a non-receptor tyrosine kinase that mediates communication in the stromal tumor microenvironment to regulate cell survival, proliferation, and migration.54 Preclinical inhibition of FAK has been shown to sensitize cells to immunotherapy and chemotherapy. A novel small molecule inhibitor of FAK, AMP945 was shown to significantly improve survival in patient derived xenograft PDAC mouse models in combination with FOLFIRINOX.55 Compared to FOLFIRINOX alone, survival was increased by 30–35% in both subcutaneous and orthotopic models. Currently, AMP945 + FOLFIRINOX is in phase Ib/IIa clinical trial for patients with unresectable or mPDAC (ACCENT Trial, NCT05355298).55
VS-6063 (defactinib) is another FAK inhibitor currently in clinical trial as an adjunct treatment. It was recently evaluated as a phase I/II clinical trial used in combination with pembrolizumab and gemcitabine in patients with advanced PDAC (NCT02546531).56 The regimen was well tolerated with no dose limiting toxicities. Twenty patients were enrolled. The DCR was 80% (one PR and 15 SD). Three patients with SD came off study due to treatment or disease-related complications. The mPFS and mOS were 5.0 and 8.3 months, respectively. Defactinib is also currently being evaluated in combination with pembrolizumab in a phase I/IIa basket trial of patients with advanced malignancies who have been offered all appropriate standard-of-care treatments (NCT02758587).
Notably, targeting another component of the desmoplastic stroma, hyaluronic acid, has not shown improvement in outcomes despite promising results in early clinical trials. Pegvorhyaluronidase alpha (PEGPH20) is a pegylated recombinant human enzyme that degrades hyaluronic acid. HALO 202 (NCT01839487), a phase II clinical trial evaluating PEGPH20 in combination with standard of care gemcitabine plus nab-paclitaxel compared to gemcitabine plus nab-paclitaxel alone in mPDAC, met its primary endpoints of mPFS and thromboembolic disease.57 mPFS was improved in the chemotherapy plus PEGPH20 arm compared to chemotherapy alone (HR 0.73, P=0.049). However, in the confirmatory phase III trial, HALO 109–301, primary endpoints of OS and PFS were not met (OS HR 1.0, PFS HR 0.97). Furthermore, a phase I/II trial of PEGHPH20 with mFOLFIRINOX backbone in mPDAC patients showed significantly worse outcomes (SWOG S1313, NCT01959139).58 PEGPH20 in combination with mFOLFIRINOX compared to mFOLFIRINOX alone in patients with mPDAC demonstrated significantly worse OS in the combination arm (PEGHPH20 + mFOLFIRINOX mOS, 7.7 months) compared to the control arm (mFOLFIRINOX, 14.4 months).
Immunotherapy
As seen in several solid tumors, microsatellite instability and deficiency in mismatch-repair (MSI-H/dMMR) have increased immunogenicity profiles and demonstrate responsiveness to ICI. Based on positive results from clinical trials that included five hundred four patients across 30 different cancers (KEYNOTE-158, KEYNOTE-164, KEYNOTE-051),59,60 pembrolizumab monotherapy was given FDA full approval for use in patients with unresectable MSI-H/dMMR solid tumors in the second line and beyond (Table 1). MSI-H/dMMR PDAC only represents 1% of PDAC61 and its use in MSI-H/dMMR is controversial. Results from KEYNOTE-158 found muted responses by PDAC to pembrolizumab compared to other non-colorectal cancers. Among twenty-two PDAC patients, the objective response rate (ORR) was only 18.2%. Other cancers demonstrated higher ORR such as endometrial (48.5%), gastric (31%), small intestine (48%), ovarian (33.3%) and biliary tract cancers (40.9%).
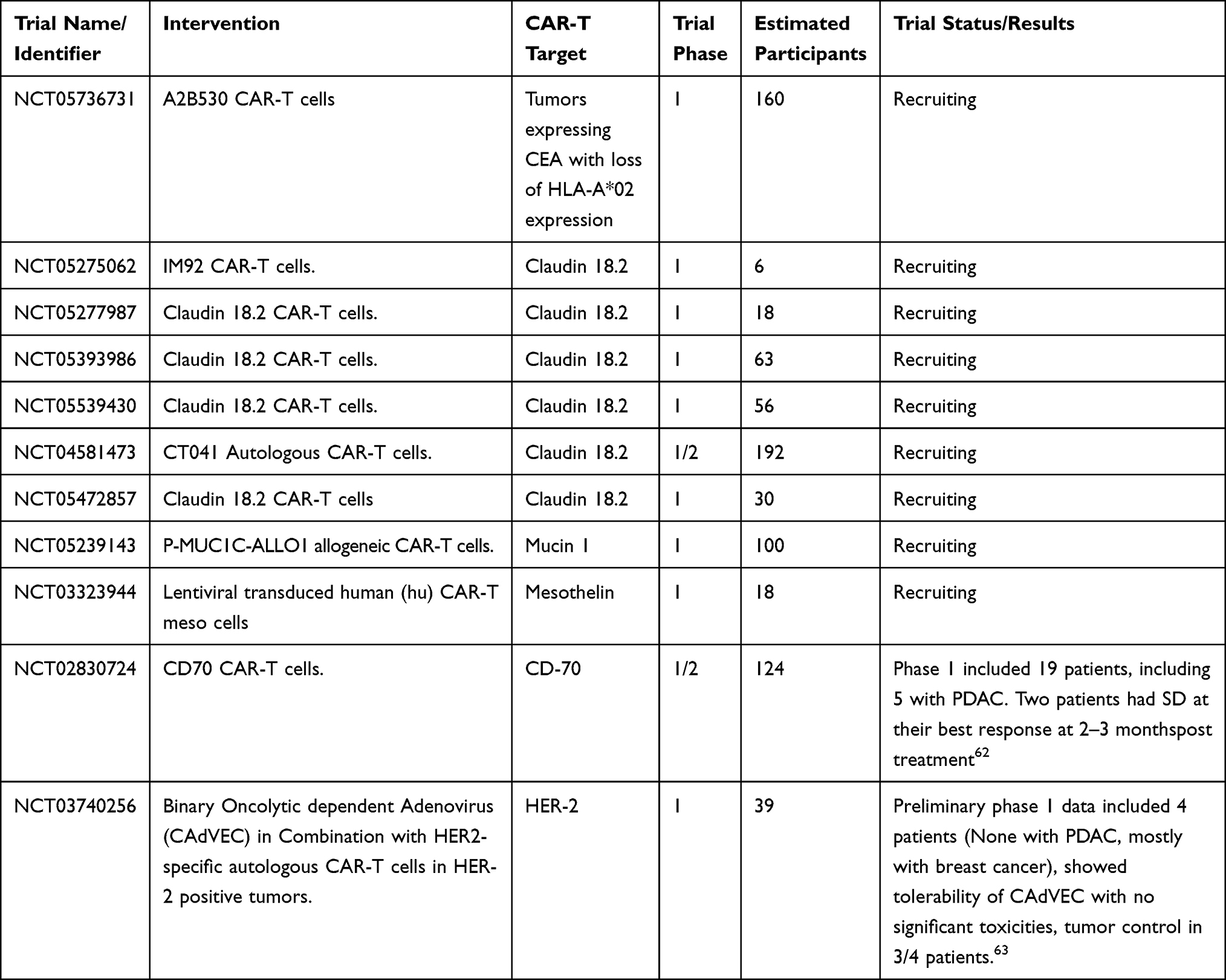 |
Table 1 Ongoing CAR-T Cell Trials in Pancreatic Cancer |
However, in retrospective studies done at Mayo Clinic and from the AGEO European Cohort, outcomes were drastically better. In the Mayo Clinic study, twenty-eight patients with MSI-H/dMMR PDAC were treated with either cytotoxic chemotherapy or immune checkpoint inhibition at Mayo Clinic. The eighteen patients that received immune checkpoint inhibition had uncharacteristically favorable responses.64 The ORR was 75% including 20% with a complete response. An additional 15% of patients had stable disease leading to a DCR of 90%. The mDOR of 14 months (5 months–93 months). The ten patients treated with systemic chemotherapy therapy had worse responses (ORR 30%, DCR 60%, mDOR 5 months). Similarly, the AGEO European Cohort study of 31 advanced MSI-H/dMMR PDAC patients treated with immune checkpoint inhibition noted an ORR of 48% and DCR of 76.5%.65 Median PFS not yet reached at a median follow-up of 18 months.
For the remaining 99% of PDAC (non MSI-H/dMMR), ICI monotherapy has failed to show any significant benefit. Additional modalities of boosting immunogenicity are needed. Targeting inflammatory cytokine signaling in combination with ICI is an area of interest. Pre-clinical data has shown a synergistic effect when IL-6 blockade is combined with immunotherapy.66 In PDAC mouse models, IL-6 blockade resulted in an increased CD8+ T cell migration into pancreatic tumors.67 This combination is currently under investigation in a phase Ib/II clinical trial in patients with previously treated PDAC (NCT04191421).68 The goal of this trial is to evaluate the safety and efficacy of siltuximab, a chimeric monoclonal antibody against IL-6 in combination with spartalizumab, a humanized IgG4 antibody against PD-1.
Another strategy involves the use of bi-specific T cell engagers (BiTE). The concept behind this strategy is to create a synapse between T cells and tumor cells with an antibody. This association leads to tumor cell killing by the attached T cell that bypasses T cell regulation and immune escape mechanisms by the tumor cells.69 ARB202 is a CDH17-CD3 BiTE antibody under investigation for gastrointestinal tumors including PDAC (NCT05411133).70 CDH17 is highly tumor-specific in gastrointestinal malignancies. It is typically limited to tight junctions of the intestine but overexpressed and location beyond tight junctions is correlated with tumor burden.71 Suppression of CDH17 in PDAC xenografts slowed tumor growth and in vitro studies showed suppression of CDH17 inhibited cell proliferation, colony formation and motility by modulating apoptosis events in PDAC cells suggesting that it may play a critical role in PDAC.
A novel approach to KRASmut PDAC is ELI-002 immunotherapy. It is a lipid-conjugated immune-stimulatory oligonucleotide (Amph-Cpg-7909) plus a mixture of lipid-conjugated peptide-based antigens (Amph-Peptides 7P).72 It acts as a lymph-node targeted therapeutic that delivers 7 KRASmut amph-peptides to enhance immunogenic response. It was studied in a multicenter phase 1 trial, AMPLIFY-201 (NCT04853017),72 to assess the safety and activity of ELI-002 monotherapy in patients with KRASmut pancreatic and colorectal cancers who have detection of minimal residual disease after first-line standard of care chemotherapy and surgery. Preliminary results presented at the 2023 ASCO Annual Meeting show that the therapy is well tolerated with no dose limiting toxicity or cytokine release syndrome associated with the treatment. Clinically, 77% of patients had biomarker reduction, and 32% achieved complete clearance. Furthermore, 87% of patients had a robust KRASmut specific T cell response. A 56-fold increased T cell response ex vivo was detected, and T cell infiltration was 10 to 29-fold higher than reported in the literature. Responses were observed at all dose levels of the therapy. A phase II trial is being planned with recommended dose of 10 mg Amph-CpG-7909.
With the success of mRNA vaccination during the COVID-19 pandemic, RNA neoantigen vaccines have come to the forefront in cancer therapeutics (NCT04486378, NCT02316457).73,74 RNA neoantigen vaccines offer a personalized approach. RNA vaccines are created from tumor samples. Gene sequencing is done on the tumor samples to identify antigens that may trigger an immune response. These genes are then used to create a personalized mRNA vaccine for the patient. A recent phase I clinical trial from Memorial Sloan Kettering Cancer Center (MSKCC) was conducted using this method was done in PDAC (NCT04161755).75 Thirty-four patients with surgically resectable untreated PDAC were enrolled in the study with sixteen patients receiving the vaccine. The study design included upfront surgery followed sequentially by a single dose atezolizumab 1200 mg IV 6 weeks after surgery, then 9 doses of vaccine (25 ug IV; 7 weekly priming doses, dose #8 at week 17 and dose #9 at week 46), then 12 cycles of mFOLFIRINOX (starting at week 21). Up to 20 neoantigens were delivered by mRNA vaccine. Patients were required to have a minimum of 5 identified antigens by next-generation sequencing to be included in the study. Therapy was well tolerated. One patient had a grade 3 adverse events (fever and hypertension) and all sixteen patients had grade 1–2 adverse events. Three patients did not receive all nine vaccine doses due to progression or death. At 18-month analysis, eight out of the sixteen treated patients (50%) demonstrated significant neo-antigen specific T cell response including long-lived polyfunctional neoantigen-specific effector CD8+ T cells. The responders clinically had longer median recurrence free survival (mRFS; not reached) than those who did not demonstrate cell response (13.4 months). Based on these results, a phase II clinical trial, IMCODE 003, evaluating the vaccine in combination with immunotherapy and chemotherapy, is currently underway. Although this study was done in resectable disease as first line therapy, it is worth noting the efficacy compared to prior vaccine trials and consideration for use in the metastatic setting.
Novel Approaches
Mesothelin is a tumor antigen that is a potential target in PDAC.76 Expression of this cell-surface glycoprotein is limited to mesothelial cells lining the pleura, peritoneum and pericardium but has also been found to be highly expressed in malignancies including PDAC where it has been found to be expressed in > 75% of PDAC.77 Its function is not completely clear, but its physiological distribution suggests that it participates in differentiation of mesothelial cells. On malignant cells, mesothelin expression correlated with poorer prognosis.
LMB-100 is a recombinant immunotoxin that contains a mesothelin-binding Fab and a Pseudomonas exotoxin A payload.78 Exotoxin A, once delivered, binds irreversibly, and modifies elongation factor-2 to stop protein synthesis. Based on preclinical data that demonstrated that the combination of LMB-100 with a taxane resulted in synergistic antitumor activity. A phase I/II study was conducted to determine safety and efficacy of LMB-100 combined with nab-paclitaxel in patients with previously treated PDAC (NCT02810418).78 Fourteen patients were enrolled, six patients received a 100 mcg/kg dose, and eight patients received a 65 mcg/kg dose. A maximum tolerated dose of 65 mcg/kg of LMB-100 (days 1, 3 and 5) combined with nab-paclitaxel (125 mg/m2 on days 1 and 8) was determined. Dose limiting toxicity from capillary leak syndrome, was seen in one of six patients receiving 65 mcg/kg dose. Response was determined by CA 19–9 levels. 7/17 patients experienced >50% decreased CA 19–9 including three patients who had prior nab-paclitaxel exposure. One patient developed an objective partial response. While this study demonstrated some activity, the dose limiting toxicity of capillary leak syndrome was prohibitive.
LMB-100 is also under investigation in combination with tofacitinib in PDAC in second line and beyond (NCT04034238). Tofacitinib is a JAK inhibitor that reduces tumor-associated inflammatory cells and has been shown to increase anti-tumor responses when used in combination with immunotoxins or antibody-drug conjugates.79
A different approach to targeting mesothelin is with chimeric antigen receptor T cells (CAR-T). CAR-T cells have been revolutionary in their applications in hematologic malignancies and are currently under investigation in multiple solid tumors (Table 1). The treatment involves harvesting a patient’s own T cells through apheresis and reprogramming them via a lentivirus vector to express a chimeric antigen receptor that is targeted towards a specific molecule. Prior to administration, the patient undergoes lymphodepletion to reduce circulating immune cell numbers allow for CAR-T cell expansion and facilitate the antitumor efficacy of the CAR-T cells.80 In this case, a CAR-T therapy directed against mesothelin (CAR-Tmeso) was given to six patients in a phase I clinical trial.81 The best response was stable disease seen in two of the six patients. These patients had PFS of 3.8 and 5.4 months, respectively. Of note, this study did not incorporate lymphodepletion prior to treatment and may have contributed to the efficacy of the treatment.
Other CAR-T treatments are in development. In a phase I clinical trial of CAR-T therapy directed against EGFR in metastatic PDAC (CAR-Tegfr), sixteen patients were enrolled82 (NCT01869166). Patients were required to have biopsy proven PDAC with EGFR expression levels of >50%. Fourteen patients were eligible for evaluation and were given a conditioning regimen that included nab-paclitaxel (100–200 mg/m2) to break down tumor stroma and cyclophosphamide (15–35 mg/m2) for lymphodepletion prior to receiving CAR-Tegfr. Four patients had a PR that lasted 2–4 months. Eight patients had SD that lasted for 2–4 months. mPFS was 3 months (2–4 months) and mOS was 4.9 months (2.9–30 months). Grade 3–4 toxicities were observed in few patients but were reversible with treatment. The most common grade 3–4 toxicity was lymphopenia, observed in 38% of patients.
Similarly, a phase I clinical trial of CAR-T therapy directed against HER2 (CAR-Ther2) in mPDAC, or metastatic biliary tract tumors (mBTC) was conducted by Feng et al (NCT01935843).83 Patients were required to have a biopsy with proven mPDAC or mBTC and expression of HER2 >50%. Two patients in the study had mPDAC. They underwent conditioning similar to CAR-Tegfr prior to the administration of CAR-Ther2. Both patients in this study achieved stable disease with a duration of response of 5.3 months and 8.3 months, respectively. Lymphopenia was the most common grade 3–4 toxicity (54%).
Another novel approach is the use of oncolytic viruses to break down the desmoplastic stroma and improve delivery of standard of care chemotherapy. VCN-01 is a novel oncolytic adenovirus genetically engineered with improved tumor targeting over its predecessors and expresses a human recombinant hyaluronidase (PH20) which facilitates intratumoral spread of the virus and breakdown of the extracellular desmoplastic stroma.84 In a phase I proof of concept study, VCN-01 was administered intratumorally to patients with PDAC in combination with chemotherapy84 (NCT02045589). Eight patients were treated with one patient having had prior treatment while the remainder did not. Inclusion criteria also included patients with negative pre-existing anti-adenovirus Ad5 levels (Nabs) which may have an impact on efficacy or on biodistribution. Although treatment was well tolerated, one patient developed massive tumor necrosis, leading to a fatal hemorrhage on day 22 after treatment. In all treated patients, the injected site was either stable or decreased in size. Out of the seven treated patients that did not experience the fatal hemorrhage, five progressed at 4 months, one progressed at 8 months and one at 31 months. Based on this serious adverse event together with the antitumor effects of VCN-01 in the tumor and the preclinical data, further study was conducted for the safety and efficacy of intravenous administration.
In a phase I/II multicenter, open label clinical trial of VCN-01 oncolytic adenovirus given intravenously with or without gemcitabine plus nab-paclitaxel with advanced solid tumors, twenty-six patients with locally advanced or metastatic, unresectable PDAC85 were included. Patients who had progressed on prior lines of therapy were included but not mandatory. 77% (20/26) of PDAC patients did not receive prior treatment while 23% (6/26) did receive prior treatment. Exploratory tumor analyses of a limited set of cases found viral replication colocalized with CD8 and inflammatory molecules such as indoleamine 2,3-dioxygenase-1 in the tumor suggesting that VCN-01 induced an inflammatory response in the tumor microenvironment. CD8 infiltration increased in 54% of the biopsies and regulatory T cells decreased in 40% of the biopsies. PD-1/PD-L1 axis was upregulated as was CTLA-4. Preliminary analysis of clinical activity from twenty-two patients found overall response rate of 50% (11/22). One patient experienced prolonged survival beyond 4 years. These results have led to first-line considerations in the ongoing phase IIb VIRAGE trial (NCT05673811). Furthermore, a phase I clinical trial is currently recruiting for the combination of VCN-01 with chimeric antigen receptor T cells targeting mesothelin in previously treated PDAC and serous epithelial ovarian cancer (NCT05057715).
Conclusion
The current standard of care treatment for unresectable/metastatic PDAC involves sequencing systemic treatment with gemcitabine- or 5-FU/leucovorin- based modalities and NAL-IRI. Modern technologies and large-scale molecular studies are improving our understanding of PDAC leading to alternative and individualized treatment strategies.
To improve current chemotherapeutic options, drug delivery systems have been evaluated. The nanoliposomal drug delivery system has enhanced intratumoral accumulation of irinotecan and its active metabolite SN-38 leading to significant improvement in outcomes and subsequent FDA approval of nanoliposomal irinotecan in the second line. At the time of this writing, it has received FDA approval for use as a first line option in combination with oxaliplatin, 5-fluorouracil and leucovorin with outcomes demonstrating superiority to gemcitabine plus nab-paclitaxel. FAK inhibitors targeting the desmoplastic stroma to improve drug delivery to the tumor and enhance efficacy of existing treatments, have been under investigation as an adjunct therapeutic strategy. Preclinical results are promising, and the inhibitors are currently in phase I/II clinical trials. Other attempts at targeting the desmoplastic stroma such as PEGPH20 did not show any benefit despite its promising preclinical results.
Alternatively, targeted therapeutics have revolutionized cancer treatment. In PDAC, targeting KRAS has remained elusive. Currently, sotorasib and adagrasib are the only KRAS targeted treatments FDA approved in cancer. Specifically, they target the KRAS G12C mutant allele which represents a small proportion of PDAC KRASmut. The most common KRAS mutation is KRAS G12D. MRTX1133 is currently in clinically trial after having shown potent in vitro and in vivo preclinical efficacy and potentially can be very promising in PDAC. Other targetable alterations are rare in PDAC.
The immunologically cold tumor microenvironment of PDAC has been problematic for immunotherapy, except in the rare cases of patients with microsatellite instability. Efforts to alter this include peptide- or mRNA- based vaccinations and CAR-T therapy. All therapies are currently under investigation in clinical trials and have shown early promising results.
Modern advances in technology and the increasing availability of these technologies are pushing the boundaries of treatment for PDAC. In this review we have highlighted multiple studies with different therapeutic strategies that are currently under investigation.
Acknowledgments
Dr. Babiker is a Paul Calabresi Scholar at the Mayo Clinic Cancer Center and acknowledges the K-12 grant program (K12CA090628). Dr. Nguyen H. Tran is a recipient of the K23MD017217-01A1 grant.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73(1):17–48. doi:10.3322/caac.21763
2. SEER Cancer Stat Facts. Cancer Stat Facts: pancreatic Cancer; 2023. Available from: https://seer.cancer.gov/statfacts/html/pancreas.html.
3. Ullman NA, Burchard PR, Dunne RF, Linehan DC. Immunologic strategies in pancreatic cancer: making cold tumors hot. J Clin Oncol. 2022;40(24):2789–2805. doi:10.1200/JCO.21.02616
4. Nakazawa H, Yoshihara S, Kudo D, et al. 4-methylumbelliferone, a hyaluronan synthase suppressor, enhances the anticancer activity of gemcitabine in human pancreatic cancer cells. Cancer Chemother Pharmacol. 2006;57(2):165–170. doi:10.1007/s00280-005-0016-5
5. Wang Y, Li XL, Mo YZ, et al. Effects of tumor metabolic microenvironment on regulatory T cells. Mol Cancer. 2018;17(1). doi:10.1186/s12943-018-0913-y
6. Rahma OE, Katz MHG, Wolpin BM, et al. Randomized multicenter phase Ib/II study of neoadjuvant chemoradiation therapy (CRT) alone or in combination with pembrolizumab in patients with resectable or borderline resectable pancreatic cancer. J Clin Oncol. 2021;39(15_suppl):4128. doi:10.1200/JCO.2021.39.15_suppl.4128
7. Gaudreau PO, Negrao MV, Mitchell KG, et al. Neoadjuvant chemotherapy increases cytotoxic T cell, tissue resident memory T cell, and B cell infiltration in resectable NSCLC. J Thorac Oncol. 2021;16(1):127–139. doi:10.1016/j.jtho.2020.09.027
8. Pelekanou V, Carvajal-Hausdorf DE, Altan M, et al. Effect of neoadjuvant chemotherapy on tumor-infiltrating lymphocytes and PD-L1 expression in breast cancer and its clinical significance. Breast Cancer Res. 2017;19(1):91. doi:10.1186/s13058-017-0884-8
9. Xing X, Shi J, Jia Y, et al. Effect of neoadjuvant chemotherapy on the immune microenvironment in gastric cancer as determined by multiplex immunofluorescence and T cell receptor repertoire analysis. J Immunother Cancer. 2022;10(3):e003984. doi:10.1136/jitc-2021-003984
10. Lim SH, Chua W, Cheng C, et al. Effect of neoadjuvant chemoradiation on tumor-infiltrating/associated lymphocytes in locally advanced rectal cancers. Anticancer Res. 2014;34(11):6505–6513.
11. Balachandran VP, Łuksza M, Zhao JN, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551(7681):512–516. doi:10.1038/nature24462
12. Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388(10039):73–85. doi:10.1016/S0140-6736(16)00141-0
13. Liu J, Ji S, Liang C, et al. Critical role of oncogenic KRAS in pancreatic cancer (Review). Mol Med Rep. 2016;13(6):4943–4949. doi:10.3892/mmr.2016.5196
14. Ischenko I, D’Amico S, Rao M, et al. KRAS drives immune evasion in a genetic model of pancreatic cancer. Nat Commun. 2021;12(1):1482. doi:10.1038/s41467-021-21736-w
15. de Leve S, Wirsdörfer F, Jendrossek V. Targeting the immunomodulatory CD73/Adenosine system to improve the therapeutic gain of radiotherapy. Front Immunol. 2019;10:698. doi:10.3389/fimmu.2019.00698
16. Zhang Z, Rohweder PJ, Ongpipattanakul C, et al. A covalent inhibitor of K-Ras(G12C) induces MHC class I presentation of haptenated peptide neoepitopes targetable by immunotherapy. Cancer Cell. 2022;40(9):1060–1069.e7. doi:10.1016/j.ccell.2022.07.005
17. Hu H, Cheng R, Wang Y, et al. Oncogenic KRAS signaling drives evasion of innate immune surveillance in lung adenocarcinoma by activating CD47. J Clin Invest. 2023;133(2). doi:10.1172/JCI153470
18. Luo J. KRAS mutation in pancreatic cancer. Semin Oncol. 2021;48(1):10–18. doi:10.1053/j.seminoncol.2021.02.003
19. Yousef A, Yousef M, Chowdhury S, et al. Impact of KRAS mutations and co-mutations on clinical outcomes in pancreatic ductal adenocarcinoma. NPJ Precis Oncol. 2024;8(1):27. doi:10.1038/s41698-024-00505-0
20. Ardalan B, Ciner A, Baca Y, et al. Not all treated KRAS- mutant pancreatic adenocarcinomas are equal: KRAS G12D and survival outcome. J Clin Oncol. 2023;41(16_suppl):4020. doi:10.1200/JCO.2023.41.16_suppl.4020
21. Luchini C, Paolino G, Mattiolo P, et al. KRAS wild-type pancreatic ductal adenocarcinoma: molecular pathology and therapeutic opportunities. J Exp Clin Cancer Res. 2020;39(1):227. doi:10.1186/s13046-020-01732-6
22. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Pancreatic adenocarcinoma V.2.2023. Available from: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1455.
23. Wang-Gillam A, Hubner RA, Siveke JT, et al. NAPOLI-1 Phase 3 study of liposomal irinotecan in metastatic pancreatic cancer: final overall survival analysis and characteristics of long-term survivors. Eur J Cancer. 2019;108:78–87. doi:10.1016/j.ejca.2018.12.007
24. Weinberg BA, Wang H, Pedersen K, Sehdev A, Sung MW, Hwang JJ. Phase II study of fluorouracil (FU), leucovorin (LV), and nanoliposomal irinotecan (nal-IRI) in previously treated advanced biliary tract cancer (NAPOLI-2). J Clin Oncol. 2020;38(4_suppl):TPS593. doi:10.1200/JCO.2020.38.4_suppl.TPS593
25. Wang-Gillam A, Li CP, Bodoky G, et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet. 2016;387(10018):545–557. doi:10.1016/S0140-6736(15)00986-1
26. Wainberg ZA, Melisi D, Macarulla T, et al. NALIRIFOX versus nab-paclitaxel and gemcitabine in treatment-naive patients with metastatic pancreatic ductal adenocarcinoma (NAPOLI 3): a randomised, open-label, phase 3 trial. Lancet. 2023;402(10409):1272–1281. doi:10.1016/S0140-6736(23)01366-1
27. Ide Y, Otsuka T, Shimokawa M, et al. Conversion surgery for unresectable pancreatic cancer treated with FOLFIRINOX or Gemcitabine Plus Nab-paclitaxel. Anticancer Res. 2023;43(4):1817–1826. doi:10.21873/anticanres.16335
28. Carrato A, Pazo-Cid R, Macarulla T, et al. Sequential nab-paclitaxel/gemcitabine followed by modified FOLFOX for first-line metastatic pancreatic cancer: the SEQUENCE trial. J Clin Oncol. 2022;40(16_suppl):4022. doi:10.1200/JCO.2022.40.16_suppl.4022
29. Kasi A, Iglesias JL. A phase 1/2 study to evaluate the safety, tolerability, and preliminary efficacy of GP-2250 in combination with gemcitabine for advanced or metastatic pancreatic adenocarcinoma. J Clin Oncol. 2022;40(4_suppl):TPS620. doi:10.1200/JCO.2022.40.4_suppl.TPS620
30. Majchrzak-Stiller B, Buchholz M, Peters I, et al. GP-2250, a novel anticancer agent, inhibits the energy metabolism, activates AMP-Kinase and impairs the NF-kB pathway in pancreatic cancer cells. J Cell Mol Med. 2023;27(14):2082–2092. doi:10.1111/jcmm.17825
31. Gwinn DM, Lee AG, Briones-Martin-Del-Campo M, et al. Oncogenic KRAS regulates amino acid homeostasis and asparagine biosynthesis via ATF4 and alters sensitivity to L-asparaginase. Cancer Cell. 2018;33(1):91–107.e6. doi:10.1016/j.ccell.2017.12.003
32. Ali U, Naveed M, Ullah A, et al. L-asparaginase as a critical component to combat Acute Lymphoblastic Leukaemia (ALL): a novel approach to target ALL. Eur J Pharmacol. 2016;771:199–210. doi:10.1016/j.ejphar.2015.12.023
33. Hammel P, Fabienne P, Mineur L, et al. Erythrocyte-encapsulated asparaginase (eryaspase) combined with chemotherapy in second-line treatment of advanced pancreatic cancer: an open-label, randomized Phase IIb trial. Eur J Cancer. 2020;124:91–101. doi:10.1016/j.ejca.2019.10.020
34. Hammel P, Feliu J, Parner V, et al. TRYbeCA-1: a randomized, phase 3 study of eryaspase in combination with chemotherapy versus chemotherapy alone as second-line treatment in patients with pancreatic adenocarcinoma (NCT03665441). Ann Oncol. 2019;30:iv26. doi:10.1093/annonc/mdz155.097
35. Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer. 2014;111(5):817–822. doi:10.1038/bjc.2014.215
36. Wang X, Allen S, Blake JF, et al. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12D inhibitor. J Med Chem. 2022;65(4):3123–3133. doi:10.1021/acs.jmedchem.1c01688
37. Strickler JH, Satake H, George TJ, et al. Sotorasib in KRAS p.G12C-mutated advanced pancreatic cancer. N Engl J Med. 2023;388(1):33–43. doi:10.1056/NEJMoa2208470
38. Bekaii-Saab TS, Spira AI, Yaeger R, et al. KRYSTAL-1: updated activity and safety of adagrasib (MRTX849) in patients (Pts) with unresectable or metastatic pancreatic cancer (PDAC) and other gastrointestinal (GI) tumors harboring a KRAS G12C mutation. J Clin Oncol. 2022;40(4_suppl):519. doi:10.1200/JCO.2022.40.4_suppl.519
39. Laskin J, Liu SV, Tolba K, et al. NRG1 fusion-driven tumors: biology, detection, and the therapeutic role of Afatinib and other ErbB-targeting agents. Ann Oncol. 2020;31(12):1693–1703. doi:10.1016/j.annonc.2020.08.2335
40. Aguirre AJ. Oncogenic NRG1 fusions: a new hope for targeted therapy in pancreatic cancer. Clin Cancer Res. 2019;25(15):4589–4591. doi:10.1158/1078-0432.CCR-19-1280
41. Fernandez-Cuesta L, Thomas RK. Molecular pathways: targeting NRG1 fusions in lung cancer. Clin Cancer Res. 2015;21(9):1989–1994. doi:10.1158/1078-0432.CCR-14-0854
42. Drilon A, Somwar R, Mangatt BP, et al. Response to ERBB3-directed targeted therapy in NRG1-rearranged cancers. Cancer Discov. 2018;8(6):686–695.
43. Rose S. MCLA-128 fights NRG1 fusion-positive cancers. Cancer Discov. 2019;9(12):1636.
44. Schram AM, Goto K, Kim DW, et al. Efficacy and safety of zenocutuzumab, a HER2 x HER3 bispecific antibody, across advanced NRG1 fusion (NRG1+) cancers. J Clin Oncol. 2022;40(16_suppl):105. doi:10.1200/JCO.2022.40.16_suppl.105
45. Kato S, Subbiah V, Marchlik E, Elkin SK, Carter JL, Kurzrock R. RET aberrations in diverse cancers: next-generation sequencing of 4871 patients. Clin Cancer Res. 2017;23(8):1988–1997. doi:10.1158/1078-0432.CCR-16-1679
46. Singhi AD, Ali SM, Lacy J, et al. Identification of targetable ALK rearrangements in pancreatic ductal adenocarcinoma. J Natl Compr Canc Netw. 2017;15(5):555–562. doi:10.6004/jnccn.2017.0058
47. Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R. The FGFR landscape in cancer: analysis of 4853 tumors by next-generation sequencing. Clin Cancer Res. 2016;22(1):259–267. doi:10.1158/1078-0432.CCR-14-3212
48. Westphalen CB, Krebs MG, Le Tourneau C, et al. Genomic context of NTRK1/2/3 fusion-positive tumours from a large real-world population. NPJ Precis Oncol. 2021;5(1):69. doi:10.1038/s41698-021-00206-y
49. Qin C, Yang G, Yang J, et al. Metabolism of pancreatic cancer: paving the way to better anticancer strategies. Mol Cancer. 2020;19(1):50. doi:10.1186/s12943-020-01169-7
50. Pająk B. Looking for the Holy Grail—drug candidates for glioblastoma multiforme chemotherapy. Biomedicines. 2022;10(5):1001. doi:10.3390/biomedicines10051001
51. Tataranni T, Agriesti F, Pacelli C, et al. Dichloroacetate affects mitochondrial function and stemness-associated properties in pancreatic cancer cell lines. Cells. 2019;8(5):478. doi:10.3390/cells8050478
52. Feuerecker B, Biechl P, Veltkamp C, Saur D, Eisenreich W. Metabolic response of pancreatic carcinoma cells under treatment with dichloroacetate. Metabolites. 2021;11(6):350. doi:10.3390/metabo11060350
53. Carbone D, De Franco M, Pecoraro C, et al. Structural manipulations of marine natural products inspire a new library of 3-Amino-1,2,4-triazine PDK inhibitors endowed with antitumor activity in pancreatic ductal adenocarcinoma. Mar Drugs. 2023;21(5):288. doi:10.3390/md21050288
54. Jiang H, Hegde S, Knolhoff BL, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22(8):851–860. doi:10.1038/nm.4123
55. Burns C, Murphy K, Cock TA, Devlin M, Herrmann D, Timpson P. The effect of adding a selective FAK inhibitor AMP945 to FOLFIRINOX in a model of pancreatic cancer. J Clin Oncol. 2023;41(16_suppl):e15128. doi:10.1200/JCO.2023.41.16_suppl.e15128
56. Wang-Gillam A, Lim KH, McWilliams R, et al. Defactinib, pembrolizumab, and gemcitabine in patients with advanced treatment refractory pancreatic cancer: a Phase I dose escalation and expansion study. Clin Cancer Res. 2022;28(24):5254–5262. doi:10.1158/1078-0432.CCR-22-0308
57. Hingorani SR, Zheng L, Bullock AJ, et al. HALO 202: randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine versus Nab-Paclitaxel/Gemcitabine in patients with untreated, metastatic pancreatic ductal adenocarcinoma. J Clin Oncol. 2018;36(4):359–366. doi:10.1200/JCO.2017.74.9564
58. Ramanathan RK, McDonough SL, Philip PA, et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX alone in patients with metastatic pancreatic adenocarcinoma: SWOG S1313. J Clin Oncol. 2019;37(13):1062–1069. doi:10.1200/JCO.18.01295
59. Diaz L, Marabelle A, Kim TW, et al. Efficacy of pembrolizumab in Phase 2 KEYNOTE-164 and KEYNOTE-158 studies of microsatellite instability high cancers. Ann Oncol. 2017;28:v128–v129. doi:10.1093/annonc/mdx367.020
60. Geoerger B, Kang HJ, Yalon-Oren M, et al. Phase 1/2 KEYNOTE-051 study of pembrolizumab (pembro) in pediatric patients (pts) with advanced melanoma or a PD-L1+advanced, relapsed, or refractory solid tumor or lymphoma. J Clin Oncol. 2017;35(15_suppl):10525. doi:10.1200/JCO.2017.35.15_suppl.10525
61. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–413. doi:10.1126/science.aan6733
62. Haas AR, Tanyi JL, O’Hara MH, et al. Phase I study of lentiviral-transduced chimeric antigen receptor-modified T cells recognizing mesothelin in advanced solid cancers. Mol Ther. 2019;27(11):1919–1929. doi:10.1016/j.ymthe.2019.07.015
63. Wang D, Porter CE, Lim B, et al. Ultralow-dose binary oncolytic/helper-dependent adenovirus promotes antitumor activity in preclinical and clinical studies. Sci Adv. 2023;9(13):eade6790. doi:10.1126/sciadv.ade6790
64. Coston T, Desai A, Babiker H, et al. Efficacy of immune checkpoint inhibition and cytotoxic chemotherapy in mismatch repair-deficient and microsatellite instability-high pancreatic cancer: Mayo Clinic experience. JCO Precis Oncol. 2023;7(7):e2200706. doi:10.1200/PO.22.00706
65. Taïeb J, Sayah L, Heinrich K, et al. Efficacy of immune checkpoint inhibitors in microsatellite unstable/mismatch repair-deficient advanced pancreatic adenocarcinoma: an AGEO European Cohort. Eur J Cancer. 2023;188:90–97. doi:10.1016/j.ejca.2023.04.012
66. Yang F, He Z, Duan H, et al. Synergistic immunotherapy of glioblastoma by dual targeting of IL-6 and CD40. Nat Commun. 2021;12(1):3424. doi:10.1038/s41467-021-23832-3
67. Mace TA, Shakya R, Pitarresi JR, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut. 2018;67(2):320–332. doi:10.1136/gutjnl-2016-311585
68. Akce M, Shaib WL, Diab M, et al. Phase Ib/II trial of siltuximab and spartalizumab in patients in metastatic pancreatic cancer. J Clin Oncol. 2022;40(4_suppl):TPS626. doi:10.1200/JCO.2022.40.4_suppl.TPS626
69. Frankel SR, Baeuerle PA. Targeting T cells to tumor cells using bispecific antibodies. Curr Opin Chem Biol. 2013;17(3):385–392. doi:10.1016/j.cbpa.2013.03.029
70. Leung RCY, Yau T, Wong DA, Luk JM, de Souza PL. Phase 1A, first-in-human study of ARB202, bispecific antibody to CDH17 and CD3, in advanced gastrointestinal malignancies expressing CDH17. JCO Glob Oncol. 2023;9(Supplement_1):25. doi:10.1200/GO.2023.9.Supplement_1.25
71. Liu X, Huang Y, Yuan H, et al. Disruption of oncogenic liver-intestine cadherin (CDH17) drives apoptotic pancreatic cancer death. Cancer Lett. 2019;454:204–214. doi:10.1016/j.canlet.2019.04.022
72. O’Reilly EM, Wainberg ZA, Weekes CD, et al. AMPLIFY-201, a first-in-human safety and efficacy trial of adjuvant ELI-002 2P immunotherapy for patients with high-relapse risk with KRAS G12D- or G12R-mutated pancreatic and colorectal cancer. J Clin Oncol. 2023;41(16_suppl):2528.
73. Schmidt M, Vogler I, Derhovanessian E, et al. 88MO T-cell responses induced by an individualized neoantigen specific immune therapy in post (neo)adjuvant patients with triple negative breast cancer. Ann Oncol. 2020;31:S276. doi:10.1016/j.annonc.2020.08.209
74. Kopetz S, Morris VK, Alonso-Orduña V, et al. A phase 2 multicenter, open-label, randomized, controlled trial in patients with stage II/III colorectal cancer who are ctDNA positive following resection to compare efficacy of autogene cevumeran versus watchful waiting. J Clin Oncol. 2022;40(16_suppl):TPS3641. doi:10.1200/JCO.2022.40.16_suppl.TPS3641
75. Rojas LA, Sethna Z, Soares KC, et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature. 2023;618(7963):144–150. doi:10.1038/s41586-023-06063-y
76. Argani P, Iacobuzio-Donahue C, Ryu B, et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin Cancer Res. 2001;7(12):3862–3868.
77. Hassan R, Thomas A, Alewine C, Le DT, Jaffee EM, Pastan I. Mesothelin immunotherapy for cancer: ready for prime time? J Clin Oncol. 2016;34(34):4171–4179. doi:10.1200/JCO.2016.68.3672
78. Alewine C, Ahmad M, Peer CJ, et al. Phase I/II study of the mesothelin-targeted immunotoxin LMB-100 with Nab-Paclitaxel for patients with advanced pancreatic adenocarcinoma. Clin Cancer Res. 2020;26(4):828–836. doi:10.1158/1078-0432.CCR-19-2586
79. Simon N, Antignani A, Hewitt SM, Gadina M, Alewine C, FitzGerald D. Tofacitinib enhances delivery of antibody-based therapeutics to tumor cells through modulation of inflammatory cells. JCI Insight. 2019;4(5). doi:10.1172/jci.insight.123281
80. Amini L, Silbert SK, Maude SL, et al. Preparing for CAR T cell therapy: patient selection, bridging therapies and lymphodepletion. Nat Rev Clin Oncol. 2022;19(5):342–355. doi:10.1038/s41571-022-00607-3
81. Beatty GL, O’Hara MH, Lacey SF, et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a Phase 1 trial. Gastroenterology. 2018;155(1):29–32. doi:10.1053/j.gastro.2018.03.029
82. Liu Y, Guo Y, Wu Z, et al. Anti-EGFR chimeric antigen receptor-modified T cells in metastatic pancreatic carcinoma: a phase I clinical trial. Cytotherapy. 2020;22(10):573–580. doi:10.1016/j.jcyt.2020.04.088
83. Feng K, Liu Y, Guo Y, et al. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell. 2018;9(10):838–847. doi:10.1007/s13238-017-0440-4
84. Bazan-Peregrino M, Garcia-Carbonero R, Laquente B, et al. VCN-01 disrupts pancreatic cancer stroma and exerts antitumor effects. J Immunother Cancer. 2021;9(11):e003254. doi:10.1136/jitc-2021-003254
85. Garcia-Carbonero R, Bazan-Peregrino M, Gil-Martín M, et al. Phase I, multicenter, open-label study of intravenous VCN-01 oncolytic adenovirus with or without nab-paclitaxel plus gemcitabine in patients with advanced solid tumors. J Immunother Cancer. 2022;10(3):e003255. doi:10.1136/jitc-2021-003255
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.