")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Spotlight on Lonapegsomatropin Once-Weekly Injection and Its Potential in the Treatment of Growth Hormone Deficiency in Pediatric Patients
Received 2 March 2022
Accepted for publication 3 June 2022
Published 29 June 2022 Volume 2022:16 Pages 2055—2066
DOI https://doi.org/10.2147/DDDT.S336285
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Bradley S Miller,1 Kevin CJ Yuen2
1Pediatric Endocrinology, University of Minnesota Medical School, Minneapolis, MN, USA; 2Department of Neuroendocrinology and Neurology, Barrow Pituitary Center, University of Arizona College of Medicine and Creighton School of Medicine, Phoenix, AZ, USA
Correspondence: Bradley S Miller, Division of Endocrinology, Department of Pediatrics, University of Minnesota Masonic Children’s Hospital, 201 Academic Office Building, 2450 Riverside Avenue S, Minneapolis, MN, 55014, USA, Tel +1 612 624 5409, Fax +1 612 626 5262, Email [email protected]
Abstract: Lonapegsomatropin, a long-acting GH therapy (LAGH), was approved by the United States Food and Drug Administration in August 2021 for the treatment of pediatric growth hormone deficiency (GHD). Lonapegsomatropin is a prodrug consisting of unmodified GH transiently conjugated to methoxypolyethylene glycol which enables time-release of GH with a half-life of ∼ 25 hours allowing for once-weekly administration. Clinical trials of lonapegsomatropin have demonstrated positive efficacy results in children (phase 2 and 3) and adults (phase 2) with GHD. The phase 3 trial in children with GHD established non-inferiority and statistical superiority of height velocity with lonapegsomatropin (11.2 cm/yr) compared to daily GH (10.3 cm/yr), with no concerning side effects with lonapegsomatropin. Similar growth responses have been reported in other LAGH products in phase 2 (somapacitan) and phase 3 (somatrogon) trials. Lonapegsomatropin is distributed in temperature-stable, prefilled cartridges at 9 different doses that can be prescribed based upon specific weight brackets designed to deliver approximately 0.24 mg/kg/wk. An electronic delivery device is required to combine the powdered medication with the diluent and deliver the medication subcutaneously through a small gauge needle to the recipient. The pharmacodynamic data from the clinical trials of lonapegsomatropin has been used to develop models to estimate an average IGF-1 value drawn at any time during the weekly injection interval. This average IGF-1 value may be used to for safety monitoring and/or to guide dose adjustment. New LAGH products, including lonapegsomatropin, may potentially improve patient adherence, quality of life and clinical outcomes, particularly in patients with poor adherence to daily GH injections in the future. With the availability of new LAGH products, clinicians will need to identify the best candidates for LAGH therapy and understand how to monitor and adjust therapy. Long-term surveillance studies are needed to demonstrate adherence, efficacy, cost-effectiveness and safety of LAGH preparations.
Keywords: growth, long-acting growth hormone, IGF-1, growth hormone deficiency, pediatric, adult
Introduction
This article describes the history of daily growth hormone (DGH) therapy, the rationale for using long-acting GH therapy (LAGH), previous attempts at generating LAGH preparations by different pharmaceutical companies, LAGH therapies currently in development, and approved LAGH therapies around the world, with a primary focus on the recently approved lonapegsomatropin. Historically, GH therapy for children with GH deficiency (GHD) began in 1958 with the use of pituitary derived human GH that was administered by intramuscular injection three times per week.1 The first attempt at developing a LAGH preparation was performed in 1979 by Lippe et al by incorporating pituitary derived human GH into gelatin to prolong its half-life.2 Subsequently, recombinant human GH (rhGH) therapy was administered daily and became available for the treatment of pediatric GHD (PGHD) in 1985 and adult GHD (AGHD) in 1996. However, because of the need for daily injections, the adherence to GH has shown to decrease over time with concomitant reductions in height velocity and insulin-like growth factor I (IGF-I) levels in the short term in children and adolescents.3 It is likely that reduced adherence to daily injections limits treatment outcomes as evidenced by adult height in children who required GH replacement therapy that are below the mean for the population.4–10
Maintenance of treatment adherence with daily GH (DGH) injections has also been shown to be problematic for adults with GHD because of device limitations, pain at injection sites, inconvenience of daily injections, lack of perceived immediate benefits, insurance barriers, and costs, leading to frequent dose omissions and treatment cessation.11,12 Thus, it has been hypothesized that LAGH products might help mitigate treatment non-adherence and potentially improve long-term treatment effects in patients with PGHD and AGHD.
Development of Long-Acting Growth Hormone Products
Nutropin Depot®, rhGH released slowly from biodegradable microspheres, was the first LAGH approved for PGHD in 1999, but was removed from the market in 2004 due to marketing and manufacturing issues. Since then, a number of other attempts have been made to develop LAGH products using different approaches to prolong the half-life of the GH molecule,13 including unmodified rhGH in a depot formulation (ie, Eutropin Plus®), pegylated rhGH (ie, Jintrolong®), modification of rhGH to increase albumin binding (ie, somapacitan, Sogroya®), rhGH fusion proteins (ie, somatrogon, NGENLA®) and prodrug releasing unmodified rhGH (ie, lonapegsomatropin, Skytrofa®). Eutropin Plus® (South Korea), Jintrolong® (China) and NGENLA® (somatrogon; Canada, Australia and Japan) are currently available for treatment of PGHD. The Phase III trial of somatrogon in AGHD (ClinicalTrials.gov Identifier: NCT01909479) was completed in 2016 and failed to meet the primary endpoint,14 whereas the other phase III trial of somatrogon in PGHD (ClinicalTrials.gov Identifier: NCT02968004) that was completed in 2019 demonstrated non-inferiority of somatrogon to DGH.15,16 Based upon these results, somatrogon received market authorization for treatment of PGHD in the European Union by the European Medicines Agency in February 2022.17 A Biologics License Application for somatrogon for the treatment of PGHD was submitted to the US Food and Drug Administration (FDA) in 2021 and received a Complete Response Letter in January 2022, but is currently not approved yet in the US.18 Sogroya® (somapacitan; US, Europe, Japan) was approved by the FDA for treatment of AGHD in August 2020, but yet to be commercially available in the US. The somapacitan Phase II trial in children with short stature associated with small for gestational age (ClinicalTrials.gov Identifier: NCT03878446) and phase III trial in PGHD (ClinicalTrials.gov Identifier: NCT03811535) were completed in 2021, but results have yet to be reported.19,20 The details of the clinical development program for lonapegsomatropin are described in Table 1, and the timeline of DGH and LAGH product availability is shown in Figure 1.
 |
Table 1 Timeline of Events in the Development of Lonapegsomatropin |
 |
Figure 1 Timeline of daily GH and long-acting GH product availability. Abbreviations: Pit-hGH, pituitary human growth hormone; GH, growth hormone; rhGH, recombinant human growth hormone; PGHD, pediatric growth hormone deficiency; AGHD, adult growth hormone deficiency; USA, United States of America; LAGH, long-acting growth hormone; EU, European Union. Notes: The timeline shows the year each GH product became available. *GH-gelatin was never approved for use. |
Mechanism of Action
Lonapegsomatropin is a prodrug that consists of native, unmodified rhGH attached covalently to an inert carrier molecule, methoxypolyethylene glycol (mPEG), via a transient linker (Figure 2). The chemical characteristics of the linker determine the pharmacokinetics and conditions of release of unmodified rhGH from the carrier prodrug. The linker present in lonapegsomatropin undergoes autohydrolysis to act as a timer to allow controlled release of GH at body temperature and pH. The prodrug is absorbed from the subcutaneous injection site into the circulation and acts as a circulatory reservoir. As GH is released from the prodrug under physiologic conditions, lonapegsomatropin predictably releases the native GH within therapeutic levels over 1 week to allow the same tissue distribution and receptor activation as endogenous GH. This includes binding to GH binding protein in the circulation and delivery to the target tissues where GH action occurs through interaction with the GH receptor. Because the prodrug releases unmodified rhGH, lonapegsomatropin is expected to be able to reach the target tissues and exert similar actions as endogenous GH. The mPEG carrier is cleared primarily by renal filtration and to a minor extent by hepatobiliary excretion.
 |
Figure 2 Schematic overview of lonapegsomatropin.24 Abbreviations: GH, growth hormone; hGH, human growth hormone; TransCon, transiently conjugated. Notes: Lonapegsomatropin is a sustained-release inactive prodrug consisting of unmodified GH transiently bound to an inert carrier via a proprietary linker. The linker undergoes predictable autohydrolysis under physiologic pH and temperature, releasing fully active GH at the GH receptor. The inert carrier is primarily cleared by renal excretion. Adapted from Thornton PS, Maniatis AK, Aghajanova E, et al. Weekly lonapegsomatropin in treatment-naive children with growth hormone deficiency: the Phase 3 heiGHt trial. J Clin Endocrinol Metab. 2021;106(11):3184–3195. Copyright © The Author(s) 2021. Published by Oxford University Press on behalf of the Endocrine Society.This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivs licence (https://creativecommons.org/licenses/by-nc-nd/4.0/).24 |
Pharmacokinetics and Pharmacodynamics
The pharmacokinetics and pharmacodynamics of lonapegsomatropin (also known as ACP-001, TransCon PEG hGH and TransCon GH) have been studied in healthy adults (Phase 1) as well as adults and children with GHD (phase 2 and 3).21–24 The absorption profile of lonapegsomatropin from the subcutaneous tissue is similar to that with DGH injections. There is a large peak of lonapegsomatropin that occurs within the first 24 hours following subcutaneous administration. Following the initial peak, there is a slow decline in GH levels over the course of one week. The half-life of lonapegsomatropin is 30.7 ± 12.7 hours, while the half-life of released GH is approximately 25 hours and the apparent clearance of lonapegsomatropin is 3.2 mL/h/kg in pediatric patients. This pharmacokinetic profile allows for weekly administration of lonapegsomatropin with no accumulation of drug over time. This contrasts to the half-life of approximately two hours for DGH after a subcutaneous injection and approximately 20 minutes after an intravenous injection.
The pharmacodynamics of lonapegsomatropin have been measured using IGF-I as the biomarker. Using IGF-I data from the phase 2 and phase 3 clinical trials in children, a pharmacodynamic model has been developed to estimate the average IGF-I SDS and average IGF-I concentrations from a single serum sample obtained at any time after an injection of lonapegsomatropin at steady state.25 It is necessary that the timing of the injection and the timing of collection of the serum sample are known in order to calculate the estimated average IGF-I level. Based upon the pharmacodynamic model, the peak IGF-I level occurs at approximately 2.5 days and the average IGF-I level occurs at approximately four days. Therefore, if a convenience sample is obtained four days after the injection, it is a reasonable estimate of the average IGF-I. If the sample is collected at any other time following the injection, the IGF-I calculator can be used to calculate the estimated average IGF-I. Because the shape of the pharmacodynamic curve should be identical regardless of the method of IGF-I assay, the IGF-I calculator could be very useful regardless of the type of IGF-I assay used. However, this calculator needs to be evaluated further in a broader population of children with GHD, including pubertal children and transition patients. Additionally, since IGF-I values are not normally distributed, the SDS values may vary by age and assay.26 For this reason, further validation of the model with different IGF-I assays are needed.
Clinical Trial Data
In clinical trials (Table 1), the dosing of lonapegsomatropin is based upon the milligrams of native GH present in the prodrug as this allows a direct comparison to dosing for daily rhGH. The first human phase 1 randomized trial published in 2017 aimed to evaluate the safety, tolerability, immunogenicity, pharmacokinetics and pharmacodynamics of a single dose of lonapegsomatropin as compared to equivalent doses of DGH (Omnitrope®) or placebo in healthy adults.22 Forty-four healthy male adults were randomized to 4 cohorts of 11 subjects, distributed in a 7:2:2 ratio (lonapegsomatropin: Omnitrope®: placebo). A single injection of 4 possible lonapegsomatropin doses (ie, 0.04, 0.08, 0.16, or 0.24 mg/kg/week) or two different Omnitrope® doses (ie, 0.08 or 0.16 mg/kg/wk divided into 7 equal daily doses) were administered and subjects were evaluated for adverse events, immunogenicity, and GH and IGF-I levels. Lonapegsomatropin was well tolerated with no injection site reactions and no anti-GH binding antibodies or electrocardiogram changes. Overall, the exposure of GH (Cmax) and IGF-I (AUC 0–168 hours) following administration of equivalent doses of lonapegsomatropin and Omnitrope® were similar, and GH and IGF-I kinetics showed a dose-proportional increase following a single administration of lonapegsomatropin. These results indicated that the prodrug is suitable for weekly administration and support its advancement to pediatric and adult GHD trials.
The phase 2 clinical trial of lonapegsomatropin compared the safety and efficacy of three different doses of once weekly lonapegsomatropin (0.14, 0.21 and 0.30 mg/kg/wk) to daily Genotropin® at a dose of 0.21 mg/kg/wk in prepubertal children with GHD over the course of 26 weeks.21 The annualized height velocity of children receiving once weekly lonapegsomatropin was 11.9 cm/yr (0.14 mg/kg/wk), 12.9 cm/yr (0.21 mg/kg/wk) and 13.9 cm/yr (0.30 mg/kg/wk) compared to 11.6 cm/yr in children receiving daily Genotropin® (0.21 mg/kg/wk). The size of this study was small with 12 to 14 children in each treatment group, the average age of the children in each group ranged from 7.5 to 8.4 years, the average height at start of treatment ranged from −2.8 to −3.3 SDS and the average peak stimulated GH ranged from 4.4 to 5.2. These characteristics demonstrate an appropriate group of children with severe growth failure related to PGHD. During this study, IGF-I levels were measured at day 7 following the injection as this represented the trough level. IGF-I levels measured during this phase 2 study were infrequently greater than +2 SDS. IGF-I levels greater than +3 SDS were only seen in the highest dose group receiving 0.30 mg/kg/wk lonapegsomatropin, were seen transiently in one subject and were not associated with any adverse events. The safety of lonapegsomatropin was comparable to daily Genotropin® with no new adverse events reported. The BMI in children receiving lonapegsomatropin was stable over the course of the study, while the BMI in children receiving daily Genotropin® decreased slightly during treatment. One subject receiving lonapegsomatropin developed anti-GH antibodies that were non-neutralizing and had no apparent effect on the growth velocity.
The phase 3 clinical trials for lonapegsomatropin included the pivotal heiGHt™ trial, a head-to-head 52 week trial comparing lonapegsomatropin (0.24 mg/kg/wk) to DGH (Genotropin® 0.24 mg/kg/wk), the fliGHt™ switch trial, a 26 week trial in which children receiving DGH for GHD transition to lonapegsomatropin, and the enliGHten™ trial which is the extension study for continued monitoring of safety and efficacy of lonapegsomatropin which children in the heiGHt™ and fliGHt™ trials transitioned into when they completed those studies.24,27,28
In the heiGHt™ trial, children were randomized 2:1 to receive lonapegsomatropin (n = 105) or daily Genotropin® (n = 56) for 52 weeks.24 The primary endpoint of the heiGHt™ trial was annualized height velocity. This trial was powered to meet the noninferiority requirements of the regulatory agencies. Based upon these requirements, lonapegsomatropin was non-inferior to DGH if the annualized height velocity in children receiving lonapegsomatropin reached a threshold of 2 cm below the annualized height velocity in children receiving daily Genotropin®. Children receiving lonapegsomatropin had an annualized height velocity of 11.2 cm/yr compared to 10.3 cm/yr in children receiving daily Genotropin®. The estimated treatment difference was 0.86 cm (P = 0.0088; 95% confidence intervals for the treatment difference 0.22–1.50). Based on these data, lonapegsomatropin was deemed to be non-inferior to DGH, and the children achieved higher annualized height velocity with lonapegsomatropin compared to daily Genotropin®. Similar growth responses to LAGH were demonstrated in the phase 2 REAL3 trial of somapacitan in PGHD with a statistically superior response to the dose (0.16 mg/kg/wk) that was chosen for the phase 3 trial and a numerically superior response to somatrogon (0.66 mg/kg/wk) in PGHD in the phase 3 trial (Table 2).15,16,29
 |
Table 2 Comparison of Growth Responses Between Long-Acting and Daily Growth Hormones in Pediatric Growth Hormone Deficiency.16,24,29 |
During the heiGHt™ trial, IGF-I levels were measured at peak (2–3 days after lonapegsomatropin injection) and trough (day 6–7 after lonapegsomatropin injection). Average IGF-I levels were >+2 SDS in 2 children receiving daily Genotropin® and did not exceed +3 SDS. In children receiving lonapegsomatropin, the estimated average IGF-I was +1.20 SDS at the peak and −0.69 SDS at trough for a change from baseline to peak of +1.89 SDS. The modeled average IGF-1 was +0.72 SDS in children receiving lonapegsomatropin. For comparison, the estimated average IGF-1 SDS in the phase 2 REAL3 trial of somapacitan in PGHD at a dose of 0.16 mg/kg/wk was +0.31 SDS (Table 2).29 Dose reductions occurred in two subjects receiving lonapegsomatropin due to asymptomatic IGF-I elevations. A dose reduction of daily Genotropin® occurred in one subject due to facial edema. There was no evidence of lonapegsomatropin accumulation or increasing IGF-I levels over time. Elevations of IGF-I levels were not associated with any adverse events. The safety of lonapegsomatropin was comparable to daily Genotropin® with no new adverse events identified. Seven subjects receiving lonapegsomatropin and two subjects receiving Genotropin® developed anti-GH antibodies that were transient, non-neutralizing and had no apparent effect on the growth velocity.
In the fliGHt™ trial, 143 children with GHD between six months and 17 years of age were recruited to be switched from DGH therapy to lonapegsomatropin at a dose of 0.24 mg/kg/wk for 26 weeks.30 Children less than three years of age could be GH-naïve. Older participants were required to be on DGH for 13–130 weeks prior to enrollment and were not required to be prepubertal. The primary endpoint of this study was safety. The youngest child enrolled was 1.2 years and three children were GH-naïve. At the time of entry into the study, the average height of the children was −1.42 SDS in the average IGF-I was +0.90 SDS. The average annualized height velocity after 26 weeks in the fliGHt™ study was 8.7 cm/yr with higher values seen in the subgroups of GH-naïve, younger children in children who had been on GH for shorter periods of time prior to transitioning to lonapegsomatropin. The average IGF-I obtained five days after lonapegsomatropin injection was +1.6 SDS. In the enliGHten™ trial, 298 children have continued to be monitored for safety and efficacy while they receive lonapegsomatropin at a dose of 0.24 mg/kg/wk. From an efficacy perspective, children who received daily Genotropin® during the heiGHt™ trial showed growth improvement when they switched to lonapegsomatropin and the group who continued lonapegsomatropin showed continued gain in height at 104 weeks.
Children who received DGH transitioned into the fliGHt™ trial and the enliGHten™ trial for a total of 78 weeks of available exposure data gained +0.73 SDS while receiving lonapegsomatropin for a height SDS at 78 weeks of −0.69 (n = 129). This demonstrates continued catch up growth while receiving lonapegsomatropin that is approaching the midparental target height of the cohort of approximately −0.3 SDS. For both the fliGHt™ and enliGHten™ trials, no new adverse events were identified, and there were no treatment emergent adverse events that led to study drug discontinuation.31
Conversely, in adults with GHD, a phase 2 multi-center, randomized, open-label, active-controlled trial designed to compare the safety (including tolerability and immunogenicity), pharmacokinetics and pharmacodynamics of three doses of weekly lonapegsomatropin to DGH (Omnitrope®) was conducted.23 Thirty-seven adult males and females with AGHD and stable on DGH therapy for at least 3 months were, following a wash-out period, randomized to one of three lonapegsomatropin doses (0.02, 0.04 and 0.08 mg/kg/week) or Omnitrope® 0.04 mg/kg/week (divided into 7 equal daily doses) for 4 weeks. Main outcomes evaluated were adverse events, immunogenicity and GH and IGF-I levels. Lonapegsomatropin was well tolerated, with fatigue and headache being the most frequent drug-related adverse events. No anti-GH binding antibodies were detected and lonapegsomatropin demonstrated a linear, dose-dependent increase in GH exposure without accumulation. GH maximum serum concentration and IGF-I exposure were similar after lonapegsomatropin or Omnitrope® administered at comparable doses. Currently, the foresiGHt™ (ClinicalTrials.gov Identifier: NCT04615273) trial is underway, which is a multicenter, phase 3, double-blind study comparing the efficacy and safety of lonapegsomatropin, placebo, and DGH for the treatment of AGHD.32 This is a 8-week dosing study and the DGH arm is included to assist clinical judgement on the trial results, and the aim is to recruit approximately 240 patients. Randomization will occur in a 1:1:1 ratio (lonapegsomatropin: placebo: DGH), and participating sites are from the US, and countries in Europe, and Asia.
Dose Adjustment
Lonapegsomatropin has been studied in clinical trials using a weight-based dosing paradigm at a dose of 0.24 mg/kg/wk.30,31,33,34 In this dosing paradigm, similar to DGH, the dose of lonapegsomatropin was adjusted for the weight of the child at specified clinical research visits. In the published data, the only other dose adjustments occurred were due to elevated IGF-I levels or adverse events. Therefore, there is little information available to guide the clinician on how to adjust the dose of lonapegsomatropin. In clinical practice, adjustment of DGH dosing has been based upon weight or body surface area, growth velocity and/or IGF-I levels. The use of IGF-I levels to guide dose adjustment of DGH therapy has been recommended for both safety and short-term efficacy purposes. From a safety perspective, it has been recommended that GH therapy increases IGF-I levels to fall within the normal range (ie, ≤ +2 SDS).35,36 From an efficacy perspective, targeting an IGF-I in the upper part of the normal range (+1 to +2 SDS) has been suggested to improve short-term efficacy.37 However, long-term efficacy of this approach has not been demonstrated. Weight-based dosing of DGH has been shown to achieve an IGF-I level close to 0 SDS depending upon the dose used.38 As described earlier, the pharmacodynamic profile of IGF-I levels following an injection of lonapegsomatropin shows an increase of IGF-I from baseline to peak about +1.9 SDS and with return to baseline before the next injection.25 It is likely that the efficacy of lonapegsomatropin will correlate with the average IGF-I level achieved.37 Therefore, it will be important to be able to estimate an average IGF-I from serum samples collected at random clinic visits. Using the IGF-I calculator to estimate average IGF-I values from these samples may help guide dose adjustment of lonapegsomatropin (Table 3).25 Lonapegsomatropin has been shown to have a linear IGF-I dose response during clinical trials suggesting that predictable changes in average IGF-I levels will be achieved with adjustments in dose of lonapegsomatropin.21,24 There have been concerns raised that short-term elevations of GH and IGF-I during LAGH therapy may be associated with short- and long-term adverse events.26,36 The GH Research Society consensus guidelines suggested that the goal of LAGH therapy is to maintain IGF-I levels within the age-appropriate range for the majority of the treatment period, as IGF-1 levels maintained within such age-appropriate range correlates with safety of treatment.36 However, peak IGF-I levels, using the pharmacodynamic model, may be able to be estimated for future analysis of their relationship to safety and efficacy. Clinicians interested in measuring peak IGF-I levels following lonapegsomatropin administration as steady-state could obtain IGF-1 measurements at approximately 2.5 days after an injection.25
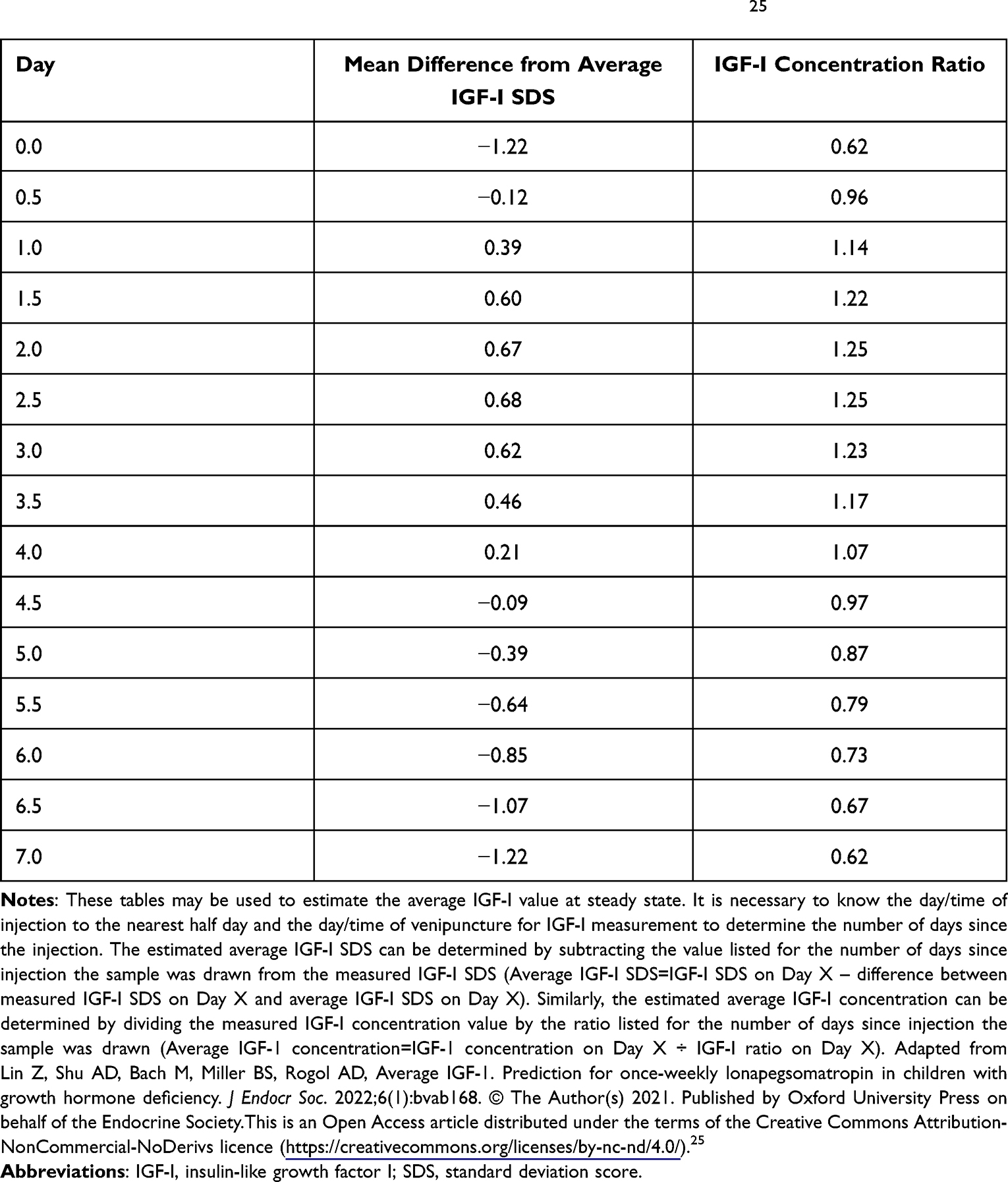 |
Table 3 The Predicted Difference Between Observed IGF-I SDS and Average IGF-I SDS and Predicted Ratio Between IGF-I and Average IGF-I Concentrations by time25 |
Current Status and Administration of Lonapegsomatropin
Lonapegsomatropin was approved by the FDA in August 2021 for the treatment of PGHD, and is marketed as Skytrofa®.39 Lonapegsomatropin is available in room temperature stable cartridges that can deliver nine different doses. The cartridges are prescribed based upon a weight range for the child with a goal of 0.24 mg/kg/wk (Table 4). For example, children weighing between 11.5 and 13.9 kg will receive a 3 mg cartridge of lonapegsomatropin. Children weighing greater than 60 kg will require two injections once per week. To administer lonapegsomatropin, the cartridges are placed into the associated electronic injection device that has a number of unique features designed to improve the injection experience for the recipient (Figure 3). The device will reconstitute the medication while prompting the user to invert the device for proper mixing. Once ready, the device will autoinject lonapegsomatropin into the subcutaneous area chosen by the parent or child. The device administers all of the medication in the cartridge with minimal waste. The electronic injection device is Bluetooth capable, but this feature is not yet approved for activation.40 In the future, if approved, the device may be able to prompt the parent or child electronically with reminders for the injection and collect injection information for tracking of adherence. The use of a bracketed dosing paradigm, where a child is matched to a cartridge based upon the weight range rather than receiving a specific amount of milligrams per kilogram per day, is a change in how pediatric endocrinologists approach the dosing of GH. This may be a beneficial change because fewer dose adjustments are needed resulting in fewer prescription changes that simplify the prescribing process for providers. However, it removes the potential to personalize the GH dose by making small dose changes based upon changes in weight, height velocity, IGF-I levels or other biomarkers. The inability to individualize therapy based upon known predictors of long-term growth responsiveness may have a negative impact on long-term effectiveness measured by adult height outcomes. In the bracketed dosing paradigm, dose adjustment would mean switching between the different size cartridges (Table 4) which results in different amounts of change based upon the weight category of the child. Therefore, children in the lower end of the weight category for each bracket will receive a dose that is higher than the recommended 0.24 mg/kg/wk and children in the higher end of the weight category for each bracket will receive a dose that is lower than recommended. It remains to be seen whether this bracketed dosing paradigm will exert a positive or negative impact on growth, adherence, ease of prescribing and side effects. Additionally, it is too early to understand how coverage of lonapegsomatropin by government payers, insurance providers and pharmacy benefit managers will impact access to this newly approved medication.
 |
Table 4 Recommended Dosing for Patients Prescribed Doses of 0.24 mg/kg/Week Based Upon Available Cartridge size39 |
 |
Figure 3 Skytrofa electronic delivery device and its features.39 Abbreviations: GH, growth hormone; mm, millimeter; SC, subcutaneous. Note: Reproduced with permission from Ascendis. SKYTROFA™ (lonapegsomatropin-tcgd) for injection, for subcutaneous use; 2021. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761177lbl.pdf. © 2022 Ascendis Pharma. All rights reserved. SKYTROFA™, Ascendis®, TransCon®, the Ascendis Pharma logo and the company logo are trademarks owned by the Ascendis Pharma Group.39 |
Patient Selection for Lonapegsomatropin
When selecting children with GHD for treatment with lonapegsomatropin, providers may consider a number of different characteristics known to negatively impact adherence. Potential candidates for lonapegsomatropin include individuals with poor adherence, particularly teenagers, young children expected to be on therapy for many years, children with needle phobia, children transitioning to self-injection and patients on multiple other medications, particularly injectable medications like insulin. Good candidates will likely be a highly motivated subset of this list of potential candidates. The prescriber needs to recognize that children with poor adherence with DGH may still have poor adherence with lonapegsomatropin. Many children, their families and their providers may be reluctant to transition to lonapegsomatropin or other LAGH products due to comfort and good growth outcomes with DGH, availability of decades of safety and efficacy data for DGH and fear of change. The cost and cost-effectiveness of lonapegsomatropin and other LAGH products is also likely to impact treatment decisions. Based upon the short-term efficacy and safety data, providers are also likely to start lonapegsomatropin in naïve children. Although lonapegsomatropin is approved for PGHD down to 1 year of age, children with severe GHD associated with hypoglycemia may not be good candidates for lonapegsomatropin or other LAGH products since they may be at increased risk of hypoglycemia at trough GH levels occurring in the day or two prior to each injection. Cancer survivors with PGHD are a group of children who warrant careful thought when considering LAGH therapy. DGH has not been shown to cause recurrent neoplasms, but concern about a small increased risk for subsequent neoplasms overall in pediatric cancer survivors remains.41 Therefore, theoretical concerns about transient elevations of GH and IGF-I that occur with each LAGH dose may lead providers to hesitate when considering LAGH therapy in cancer survivors with PGHD.42 As more safety and efficacy data for LAGH emerges and as experience with lonapegsomatropin and other LAGH products grows, it is possible that LAGH may potentially replace DGH in the treatment of PGHD.
Conclusion
Numerous LAGH preparations have been or are currently being developed but they all have their unique molecular characteristics and clinical efficacies.13 In terms of lonapegsomatropin, it has been found to be non-inferior to and statistically superior to DGH, with comparable safety in PGHD, and may potentially improve patient adherence, quality of life and clinical outcomes, particularly in patients with poor adherence to DGH injections.
Future Directions
Although a transition from daily injections to once weekly injections has been shown to improve adherence in other treatment areas, it has not yet been demonstrated in children receiving LAGH preparations such as lonapegsomatropin.43–46 It will be important to document improved adherence through standard methodologies including pharmacy refill data. If the Bluetooth capability of the electronic injection device for lonapegsomatropin is approved, this will provide additional information to correlate adherence with treatment outcomes. Although LAGH preparations are being evaluated through a regulatory process that requires demonstration of non-inferiority to DGH injections, it is likely that these compounds will result in improved long-term efficacy as well as convenience for patients and their caregivers. This improvement in outcomes will likely be due to the underestimated impact of reduced adherence and persistence with GH therapy. Long-term studies, however, are still needed to demonstrate these benefits as they are crucial in determining the cost-effectiveness and safety of LAGH preparations.
Neutralizing antibodies were not identified in lonapegsomatropin clinical trials. However, the modifications of the native GH molecule to prolong the half-life of the various LAGH products and their different pharmacokinetic profiles may activate the immune response leading to an increase in the development of neutralizing antibodies over time. Therefore, surveillance for the development of anti-LAGH antibodies may need to continue over the long term.
Long-term safety of lonapegsomatropin also requires further study as it does not mimic the physiologic profile of endogenous GH secretion, although it could be argued that DGH administration also does not have a physiologic profile either. It remains to be seen whether this difference in pharmacokinetic and pharmacodynamic profile will exert a positive or negative impact on short- and long-term safety and efficacy. Following the approval of rhGH in 1985, numerous Phase 4 postmarketing surveillance registries have collected safety and efficacy data for DGH therapy.47 These studies accumulated more than 500,000 patient years of safety and efficacy data and helped our community learn about common and rare side effects of DGH therapy. One of the challenges of these studies were that children were no longer followed after DGH therapy was discontinued, and were therefore lost to follow-up. Herculean efforts have been performed in Europe to collect retrospective data in patients who received DGH therapy in the 1980s and 1990s resulting in the landmark SAGhE study.48–50 In order to collect important data regarding linear growth and metabolic outcomes in children receiving LAGH preparations, including lonapegsomatropin, it will be crucial to perform similar phase 4 postmarketing surveillance studies. However, in order to avoid losing the patients when they complete therapy or transition to another GH product and to capture patient reported outcomes, it is imperative to develop a study utilizing LAGH therapy that follows children long-term well into their adulthood. Efforts to develop an international study to achieve these outcomes is currently underway and needs to be supported by the manufacturers of DGH and LAGH preparations as well as the pediatric endocrinology community.
Disclosure
Dr. Miller is a consultant for AbbVie, Ascendis Pharma, BioMarin, Bristol Myers Squibb, EMD Serono, Endo Pharmaceuticals, Novo Nordisk, Orchard Therapeutics, Pfizer, Tolmar and Vertice and has received research support from Alexion, AbbVie, Aeterna Zentaris, Amgen, Amicus, Lumos Pharma, Lysogene, Novo Nordisk, OPKO Health Pfizer, Prevail Therapeutics and Sangamo Therapeutics.
Dr. Yuen has served as an occasional advisory board member for Novo Nordisk, Ascendis, Sandoz, Corcept, Novartis, Ipsen, Amryt, Strongbridge, Crinetics and Recordati and has received research grants to Barrow Neurological Institute from Ascendis, Corcept, Amryt, and Novartis. The authors report no other conflicts of interest in this work.
References
1. Raben MS. Treatment of a pituitary dwarf with human growth hormone. J Clin Endocrinol Metab. 1958;18(8):901–903. doi:10.1210/jcem-18-8-901
2. Lippe B, Frasier SD, Kaplan SA. Use of growth hormone-gel. Arch Dis Child. 1979;54(8):609–613. doi:10.1136/adc.54.8.609
3. De Pedro S, Murillo M, Salinas I, et al. Variability in adherence to rhGH treatment: socioeconomic causes and effect on children’s growth. Growth Horm Igf Res. 2016;26:32–35. doi:10.1016/j.ghir.2015.12.002
4. August GP, Julius JR, Blethen SL. Adult height in children with growth hormone deficiency who are treated with biosynthetic growth hormone: the National Cooperative Growth Study experience. Pediatrics. 1998;102(Supplement_3):512–516. doi:10.1542/peds.102.S3.512
5. Bell JJ, Lippe B, Romano AA, Cernich JT, Swinford RD, Moawad D. National cooperative growth study: 25 years of growth hormone data, insights, and lessons for future registries. Pediatr Endocrinol Rev. 2018;16(2):240–255. doi:10.17458/per.vol16.2018.25yearsghdata
6. Child CJ, Zimmermann AG, Chrousos GP, et al. Safety outcomes during pediatric GH therapy: final results from the prospective genesis observational program. J Clin Endocrinol Metab. 2019;104(2):379–389. doi:10.1210/jc.2018-01189
7. Pfaffle R, Bidlingmaier M, Kreitschmann-Andermahr I, et al. Safety and effectiveness of omnitrope r, a biosimilar recombinant human growth hormone: more than 10 years’ experience from the PATRO Children Study. Horm Res Paediatr. 2020;93(3):154–163. doi:10.1159/000508190
8. Reiter EO, Price DA, Wilton P, Albertsson-Wikland K, Ranke MB. Effect of growth hormone (GH) treatment on the near-final height of 1258 patients with idiopathic GH deficiency: analysis of a large international database. J Clin Endocrinol Metab. 2006;91(6):2047–2054. doi:10.1210/jc.2005-2284
9. Ross JL, Lee PA, Gut R, Germak J. Attaining genetic height potential: analysis of height outcomes from the ANSWER Program in children treated with growth hormone over 5 years. Growth Horm Igf Res. 2015;25(6):286–293. doi:10.1016/j.ghir.2015.08.006
10. Miller BS, Rose SR, Ross JL, et al. Growth hormone dosage associated with change in height standard deviation score over 4 years of treatment by age in pediatric patients with Isolated Growth Hormone Deficiency (IGHD): results from the ANSWER program.
11. Auer MK, Stieg MR, Hoffmann J, Stalla GK. Is insulin-like growth factor-I a good marker for treatment adherence in growth hormone deficiency in adulthood? Clin Endocrinol (Oxf). 2016;84(6):862–869. doi:10.1111/cen.13030
12. Mancini A, Vergani E, Bruno C, Palladino A, Brunetti A. Relevance of adherence monitoring in adult patients with growth hormone deficiency under replacement therapy: preliminary monocentric data with easypodtm connect. Front Endocrinol. 2019;10:416. doi:10.3389/fendo.2019.00416
13. Miller BS, Velazquez E, Yuen KCJ. Long-acting growth hormone preparations - current status and future considerations. J Clin Endocrinol Metab. 2020;105(6):01. doi:10.1210/clinem/dgz149
14. Strasburger CJ, Vanuga P, Payer J, et al. MOD-4023, a long-acting carboxy-terminal peptide-modified human growth hormone: results of a Phase 2 study in growth hormone-deficient adults. Eur J Endocrinol. 2017;176(3):283–294. doi:10.1530/EJE-16-0748
15. Deal CL, Pastrak A, Silverman LA, Valluri SR, Wajnrajch MP, Cara JF. OR10-06 somatrogon growth hormone in the treatment of pediatric growth hormone deficiency: results of the pivotal pediatric Phase 3 clinical trial. J Endocr Soc. 2020;4(Supplement_1). doi:10.1210/jendso/bvaa046.1279
16. Pfizer. OPKO and pfizer announce positive Phase 3 top-line results for somatrogon, an investigational long-acting human growth hormone to treat children with growth hormone deficiency; 2019. Available from: https://www.pfizer.com/news/press-release/press-release-detail/opko_and_pfizer_announce_positive_phase_3_top_line_results_for_somatrogon_an_investigational_long_acting_human_growth_hormone_to_treat_children_with_growth_hormone_deficiency.
17. Pfizer. Pfizer and OPKO’s once-weekly NGENLA™ (somatrogon) injection receives marketing authorization in European Union for treatment of pediatric growth hormone deficiency; 2022. Available from: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-and-opkos-once-weekly-ngenlatm-somatrogon-injection.
18. Park B. FDA denies approval of long-acting pediatric growth hormone therapy; 2021. Available from: https://www.empr.com/home/news/drugs-in-The-pipeline/fda-denies-approval-of-long-acting-pediatric-growth-hormone-therapy/.
19. ClinicalTrials.gov. A research study in children born small and who stayed small. treatment is somapacitan once a week compared to Norditropin® once a day; 2019. Available from: https://clinicaltrials.gov/ct2/show/NCT03878446.
20. ClinicalTrials.gov. A research study in children with a low level of hormone to grow. treatment is somapacitan once a week compared to Norditropin® once a day (REAL4); 2019. Available from: https://clinicaltrials.gov/ct2/show/NCT03811535.
21. Chatelain P, Malievskiy O, Radziuk K, et al. A randomized Phase 2 study of long-acting TransCon GH vs daily GH in childhood GH deficiency. J Clin Endocrinol Metab. 2017;102(5):1673–1682. doi:10.1210/jc.2016-3776
22. Gilfoyle D, Mortensen E, Christoffersen ED, Leff JA, Beckert M. A first-in-man phase 1 trial for long-acting TransCon growth hormone. Growth Horm Igf Res. 2018;39:34–39. doi:10.1016/j.ghir.2017.12.002
23. Hoybye C, Pfeiffer AF, Ferone D, et al. A phase 2 trial of long-acting TransCon growth hormone in adult GH deficiency. Endocr Connect. 2017;6(3):129–138. doi:10.1530/EC-17-0007
24. Thornton PS, Maniatis AK, Aghajanova E, et al. Weekly lonapegsomatropin in treatment-naive children with growth hormone deficiency: the Phase 3 heiGHt trial. J Clin Endocrinol Metab. 2021;106(11):3184–3195. doi:10.1210/clinem/dgab529
25. Lin Z, Shu AD, Bach M, Miller BS, Rogol AD, Average IGF-1. Prediction for once-weekly lonapegsomatropin in children with growth hormone deficiency. J Endocr Soc. 2022;6(1):bvab168. doi:10.1210/jendso/bvab168
26. Bidlingmaier M, Schilbach K. The use of IGF-I to monitor long-acting growth hormone therapy-timing is an art. J Clin Endocrinol Metab. 2021;106(5):e2367–e2369. doi:10.1210/clinem/dgab016.
27. ClinicalTrials.gov. A safety, tolerability and efficacy study of TransCon hGH in children with growth hormone deficiency; 2017. Available from: https://clinicaltrials.gov/ct2/show/NCT03305016.
28. ClinicalTrials.gov. A long-term trial investigating safety and efficacy of TransCon hGH in children with growth hormone deficiency who have completed a prior TransCon hGH clinical trial (enliGHten); 2017. Available from: https://clinicaltrials.gov/ct2/show/NCT03344458.
29. Savendahl L, Battelino T, Brod M, et al. Once-weekly somapacitan vs daily GH in children with GH deficiency: results from a randomized Phase 2 trial. J Clin Endocrinol Metab. 2020;105(4):01. doi:10.1210/clinem/dgz310
30. Maniatis AK, Nadgir U, Saenger P, et al. OR10-05 Phase 3 FliGHt trial: experience of switching from daily growth hormone therapy to once-weekly TransCon HGH in children with growth hormone deficiency. J Endocr Soc. 2020;4(Supplement_1). doi:10.1210/jendso/bvaa046.963
31. Maniatis AK, Nadgir U, Hofman P, et al. Poster 154: LONAPEGSOMATROPIN (TransCon hGH) in children with growth hormone deficiency: efficacy and safety of up to 2 years of treatment. Horm Res Paediatr. 2021;94:1–203.
32. ClinicalTrials.gov. A trial to compare the efficacy and safety of once-weekly lonapegsomatropin with placebo and a daily somatropin product in adults with growth hormone deficiency (foresiGHt); 2020. Available from: https://clinicaltrials.gov/ct2/show/NCT04615273.
33. Maniatis AK, Casella SJ, Nadgir UM, et al. Efficacy and safety of up to 2 years of treatment with TransCon hGH (Lonapegsomatropin) in treatment-naïve and treatment-experienced children with growth hormone deficiency. J Endocr Soc. 2021;5(Supplement_1):A676–A676. doi:10.1210/jendso/bvab048.1378
34. Thornton P, Hofman P, Maniatis AK, et al. TransCon growth hormone in the treatment of pediatric growth hormone deficiency results of the Phase 3 HeiGHt trial; 2019. https://ascendispharma.com/wp-content/uploads/ENDO-2019-heiGHt-Trial-Presentation.pdf.
35. Allen DB, Backeljauw P, Bidlingmaier M, et al. GH safety workshop position paper: a critical appraisal of recombinant human GH therapy in children and adults. Eur J Endocrinol. 2016;174(2):P1–P9. doi:10.1530/EJE-15-0873
36. Christiansen JS, Backeljauw PF, Bidlingmaier M, et al. Growth hormone research society perspective on the development of long-acting growth hormone preparations. Eur J Endocrinol. 2016;174(6):C1–C8. doi:10.1530/EJE-16-0111
37. Park P, Cohen P. The role of insulin-like growth factor I monitoring in growth hormone-treated children. Horm Res. 2004;62(Suppl 1):59–65. doi:10.1159/000080760
38. Cohen P, Rogol AD, Howard CP, et al. Insulin growth factor-based dosing of growth hormone therapy in children: a randomized, controlled study. J Clin Endocrinol Metab. 2007;92(7):2480–2486. doi:10.1210/jc.2007-0204
39. Ascendis. SKYTROFA™ (lonapegsomatropin-tcgd) for injection, for subcutaneous use; 2021. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761177lbl.pdf.
40. Walsh K, Campbell D, Permuy J, et al. Introduction of a novel GH auto-injector for once-weekly administration of TransCon hGH.
41. Raman S, Grimberg A, Waguespack SG, et al. Risk of neoplasia in pediatric patients receiving growth hormone therapy–A report from the pediatric endocrine society drug and therapeutics committee. J Clin Endocrinol Metab. 2015;100(6):2192–2203. doi:10.1210/jc.2015-1002
42. Allen DB, Merchant N, Miller BS, Backeljauw PF. Evolution and future of growth plate therapeutics. Horm Res Paediatr. 2021;94(9–10):319–332. doi:10.1159/000520812
43. Djambas Khayat C. Once-weekly prophylactic dosing of recombinant factor IX improves adherence in hemophilia B. J Blood Med. 2016;7:275–282. doi:10.2147/JBM.S84597
44. Jose B, Tahrani AA, Piya MK, Barnett AH. Exenatide once weekly: clinical outcomes and patient satisfaction. Patient Prefer Adherence. 2010;4:313–324.
45. Li A, Goodfriend C, Sokol J, Kruse-Jarres R. Patterns and predictors of emicizumab adherence in people with hemophilia. Blood. 2019;134(Supplement_1):2178. doi:10.1182/blood-2019-128083
46. Qiao Q, Ouwens MJ, Grandy S, Johnsson K, Kostev K. Adherence to GLP-1 receptor agonist therapy administered by once-daily or once-weekly injection in patients with type 2 diabetes in Germany. Diabetes Metab Syndr Obes Targets Ther. 2016;9:201–205. doi:10.2147/DMSO.S99732
47. Miller BS, Rosenfeld RG. Monitoring rhGH safety: rhGH registries, SAGhE and future needs. Pediatr Endocrinol Rev. 2018;16(Suppl 1):150–161. doi:10.17458/per.vol16.2018.mr.monitoringrhghsafety
48. Savendahl L, Cooke R, Tidblad A, et al. Long-term mortality after childhood growth hormone treatment: the SAGhE cohort study. Lancet Diabetes Endocrinol. 2020;8(8):683–692. doi:10.1016/S2213-8587(20)30163-7
49. Swerdlow AJ, Cooke R, Albertsson-Wikland K, et al. Description of the SAGhE cohort: a large European study of mortality and cancer incidence risks after childhood treatment with recombinant growth hormone. Horm Res Paediatr. 2015;84(3):172–183. doi:10.1159/000435856
50. Swerdlow AJ, Cooke R, Beckers D, et al. Cancer risks in patients treated with growth hormone in childhood: the SAGhE European cohort study. J Clin Endocrinol Metab. 2017;102(5):1661–1672. doi:10.1210/jc.2016-2046
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.