")
Back to Journals » Journal of Blood Medicine » Volume 14
Switching from Sucrose-Formulated rFVIII to Octocog Alfa (BAY 81-8973) Prophylaxis Improves Bleed Outcomes in the LEOPOLD Clinical Trials
Authors Kenet G, Moulton T, Wicklund BM , Ahuja SP, Escobar M , Mahlangu J
Received 21 January 2023
Accepted for publication 18 May 2023
Published 7 June 2023 Volume 2023:14 Pages 379—388
DOI https://doi.org/10.2147/JBM.S405624
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Gili Kenet,1,2 Thomas Moulton,3 Brian M Wicklund,4 Sanjay P Ahuja,5 Miguel Escobar,6 Johnny Mahlangu7
1National Hemophilia Center, Sheba Medical Center, Tel HaShomer, Israel; 2The Amalia Biron Thrombosis Research Institute, Tel Aviv University, Tel Aviv, Israel; 3Bayer, Whippany, NJ, USA; 4Children’s Mercy-Kansas City, Kansas City, MO, USA; 5Rainbow Babies & Children’s Hospital, Cleveland, OH, USA; 6University of Texas Health Science Center, Houston, TX, USA; 7Hemophilia Comprehensive Care Center, Faculty of Health Sciences, Charlotte Maxeke Johannesburg Academic Hospital, University of the Witwatersrand, and National Health Laboratory Service, Johannesburg, South Africa
Correspondence: Gili Kenet, Amalia Biron Thrombosis Research Institute, Tel Aviv University, Tel HaShomer, 52621, Israel, Tel +972-3-5307356, Fax +972-3-5351806, Email [email protected]
Introduction: Previous clinical trials established the efficacy and safety of sucrose-formulated recombinant factor (F) VIII (rFVIII-FS/Kogenate FS®/Helixate FS®) and octocog alfa (BAY 81– 8973/Kovaltry®; LEOPOLD trials).
Aim: To report the results of a post hoc subgroup analysis assessing efficacy and safety outcomes in patients with hemophilia A who were receiving rFVIII-FS prior to enrolling into the LEOPOLD I Part B and LEOPOLD Kids Part A clinical trials and switching to octocog alfa.
Methods: LEOPOLD I Part B (NCT01029340) and LEOPOLD Kids Part A (NCT01311648) were octocog alfa Phase 3, multinational, open-label studies in patients with severe hemophilia A aged 12– 65 years and ≤ 12 years, respectively. Annualized bleeding rate (ABR) was the efficacy endpoint for both studies. Safety endpoints included adverse events (AEs) and development of FVIII inhibitors.
Results: Of the 113 patients in both LEOPOLD trials, 40 (35.4%) patients received rFVIII-FS prophylaxis pre-study and had data available for pre-study total ABR. In LEOPOLD I Part B (n = 22, 35.5%), median (Q1; Q3) total ABR decreased from 2.5 (0.0; 9.0) pre-study to 1.0 (0.0; 6.8), and from 1.0 (0.0; 6.0) pre-study to 0.0 (0.0; 6.02) in LEOPOLD Kids Part A (n = 18, 35.3%). Octocog alfa was well tolerated, and no patients had drug-related serious AEs or inhibitors.
Conclusion: Treatment with octocog alfa prophylaxis appeared to have a favorable risk–benefit profile compared with rFVIII-FS and thus could be an effective and improved alternative strategy for individualized treatment for children, adolescent and adult patients with severe hemophilia A currently on rFVIII-FS treatment.
Keywords: FVIII, hemophilia A, prophylaxis, recombinant proteins, octocog alfa
Introduction
Hemophilia A is caused by impaired thrombin generation due to an absence or deficiency of factor VIII (FVIII).1 Regular replacement therapy with FVIII products, known as prophylaxis, is considered the standard of care.1 For patients receiving their initial therapy, factors including efficacy, safety, cost and supply/availability might determine the choice of FVIII product.2,3 There are multiple drivers to switch treatment products in patients with hemophilia A, including national contracting, access to products and patient preference. The key reasons for switching to an alternative treatment are the clinical benefits for patients, including improved efficacy or pharmacokinetic (PK) profile, thus providing an overall better treatment outcome than the current FVIII treatment. As a result of the evolving treatment landscape in hemophilia, it is unusual for adult patients to use the same FVIII concentrate throughout their lives.3 There are, however, both patient and physician barriers to switching and often a reluctance to change products. These barriers include concerns of perceived possible negative outcomes, such as increased risk of FVIII-inhibitor development and strong patient preference to continue their current therapy, often due to the development of a strong psychological link with the product they use.2,3
Sucrose-formulated recombinant FVIII (rFVIII-FS, Kogenate FS®/Helixate FS®) and octocog alfa (BAY 81–8973, Kovaltry®) are unmodified, full-length, recombinant FVIII proteins, indicated for prophylaxis and treatment of bleeds in children and adults with hemophilia A since 1993 and 2016, respectively.4,5 These approvals were based on the results from previous clinical trials demonstrating the efficacy and safety of on-demand and prophylactic treatment with rFVIII-FS (including the SPINART trial) and octocog alfa (LEOPOLD trials) in patients with hemophilia A.6–14 While rFVIII-FS and octocog alfa are full-length human rFVIII products that share the same primary amino acid sequence, these products differ in the way they are manufactured. No human or animal proteins are used in the final formulation steps during the manufacturing of rFVIII-FS, unlike its predecessor rFVIII product, although the cell culture medium has human plasma protein in the solution.15 Octocog alfa, on the other hand, contains no human or animal-derived raw materials added to the cell culture, purification or formulation processes.15–17 In addition, improved manufacturing methodologies including viral filtration, enhanced viability of the expression cell line using heat shock protein 70 and ultra-fast membrane-based capture technology resulted in a rFVIII product with high and consistent purity. Moreover, octocog alfa has key structural changes such as increased branching and sialylation on N-terminal glycan groups compared with its predecessors, known to be critical to the half-life of some mammalian proteins.15–17
PK assessments based on both one-stage clotting and chromogenic assays indicated a favorable PK profile for octocog alfa with a higher area under the curve and half-life, and a markedly lower clearance compared with rFVIII-FS.15 The observed favorable PK profile for octocog alfa compared with rFVIII-FS was noted across all age groups, particularly in the pediatric age group, and a high proportion of adults and adolescent patients achieved sustained FVIII levels ≥1%.11,12,15 Octocog alfa also demonstrated a superior PK profile compared with antihemophilic factor (recombinant) plasma/albumin-free method (rAHF-PFM/Advate®) in a randomized cross-over head-to-head comparison.18 These PK studies provided rationale for exploring the efficacy of switching to octocog alfa in previously FVIII-treated patients.
The efficacy and safety of octocog alfa in previously treated patients were assessed in the phase 3 LEOPOLD I Part B and LEOPOLD Kids Part A trials.11,12 Upon study entry, patients switched from their previous FVIII treatment to octocog alfa. Here, we report the results of a post hoc subgroup analysis that aimed to assess the efficacy and safety outcomes of patients with hemophilia A who were receiving rFVIII-FS prior to enrolling into the LEOPOLD I Part B and LEOPOLD Kids Part A clinical trials and subsequently switched to octocog alfa upon study entry.
Materials and Methods
Patients and Study Design
The eligibility criteria and study designs of the LEOPOLD I Part B and LEOPOLD Kids Part A trials have been described previously.11,12 Briefly, LEOPOLD I Part B and LEOPOLD Kids Part A were phase 3, multinational, open-label studies in patients with severe hemophilia A (ClinicalTrials.gov identifiers: NCT01029340 and NCT01311648, respectively).
In LEOPOLD I Part B, males aged 12–65 years with severe hemophilia A (FVIII <1%) previously treated with any FVIII product for ≥150 exposure days (EDs) were eligible for inclusion.11 In LEOPOLD Kids Part A, patients aged ≤12 years with severe hemophilia A (FVIII <1%) previously treated with any FVIII product for ≥50 EDs were eligible for inclusion.12 In both the LEOPOLD trials, key exclusion criteria included the presence or history of FVIII inhibitors, diagnosis of any bleeding disorder other than hemophilia A, platelet count <100,000/mm3, abnormal renal function or clinically relevant liver disease.
In LEOPOLD I Part B, patients received octocog alfa prophylaxis as intravenous administrations of 20–50 IU/kg twice or three times weekly, with the nominal dose and dosing frequency maintained throughout the study (Figure 1). Patients were randomized 1:1 to receive octocog alfa with potency of the dose determined by the chromogenic substrate assay per European Pharmacopoeia (CS/EP) or adjusted by a predefined factor to mimic results obtained with the one-stage assay (CS/ADJ) for a 6-month treatment period. Patients then crossed over to the alternate potency for another 6-month treatment period. The primary efficacy endpoint for LEOPOLD I Part B was annualized bleeding rate (ABR; defined as spontaneous and trauma-related bleeds, untreated bleeds and unspecified bleeds) in each 6-month potency period and over the whole year of treatment, independent of potency.11
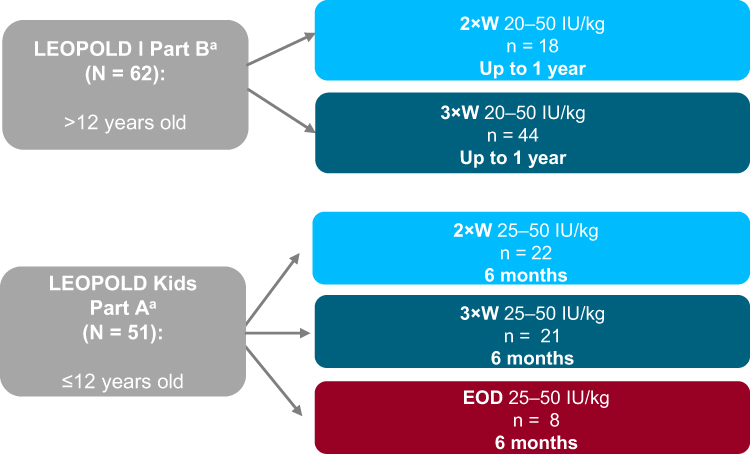 |
Figure 1 Patient disposition in the LEOPOLD trials. aDosing regimens for both studies were assigned by the investigator. Abbreviations: 2×W, twice weekly; 3×W, three times weekly, EOD, every other day. |
In LEOPOLD Kids Part A, patients received 20–50 IU/kg octocog alfa prophylaxis for ≥2 times weekly as determined by the investigator in conjunction with the patient/caregiver (Figure 1). The dose of octocog alfa infusions could be changed at any time. The primary efficacy endpoint was ABR occurring within 48 hours after prophylaxis infusion.
Bleeding events and administered infusions were recorded by patients and parents/caregivers using an electronic patient diary, during the LEOPOLD I Part B and LEOPOLD Kids Part A trials, respectively. ABRs were calculated for all bleeds (sum of spontaneous bleeds, trauma bleeds, untreated bleeds and unspecified bleeds) as well as joint, spontaneous, and trauma-related bleeds throughout each study. Pre-study ABRs were calculated using self-reported data based on patient/carer recall of the bleeding events at the baseline visit. Throughout the study, patients were closely monitored at each study visit for the incidence of adverse events (AEs) and also immunogenicity, including FVIII inhibitor development. Inhibitor development was defined as a Nijmegen-modified Bethesda assay measured titre of ≥0.6 BU and confirmed in a second plasma sample.
Both studies were approved by the independent ethics committee or institutional review board responsible for each site and were carried out in compliance with the protocol, the principles of the Declaration of Helsinki, and the International Conference on Harmonization guidelines for Good Clinical Practice. All patients or their guardians provided written informed consent. As this is a post hoc subgroup analysis of previously reported studies that had each received ethical approval, additional approval was not sought.
Statistical Analyses
Statistical analysis was performed using SAS software 9.2 (SAS Institute Inc., Cary, NC, USA). Summary statistics were calculated for continuous data, and frequencies were calculated for categorical data. Analyses performed in this study were descriptive. Further details on statistical analyses performed in the main studies are described in previous publications.11,12
Results
Patients
Out of a total of 113 patients who were treated with octocog alfa in LEOPOLD I Part B and LEOPOLD Kids Part A, a total of 40 (35.4%) patients previously received rFVIII-FS prophylaxis treatment prior to enrollment into the respective LEOPOLD trials and were included in this analysis. Pre-study total ABR data were available for all 40 patients. Disease characteristics and patient demographics at baseline can be found in Table 1. In LEOPOLD I Part B, 22 (35.5%) patients were previously treated with rFVIII-FS prophylaxis and had a median age of 27.0 years; in LEOPOLD Kids Part A, 18 (35.3%) patients were previously treated with rFVIII-FS prophylaxis and had a median age of 5.0 years (Table 1). In general, these switch cohorts had similar patient demographics to the overall study population (Table 1), and any differences did not affect the final analysis.
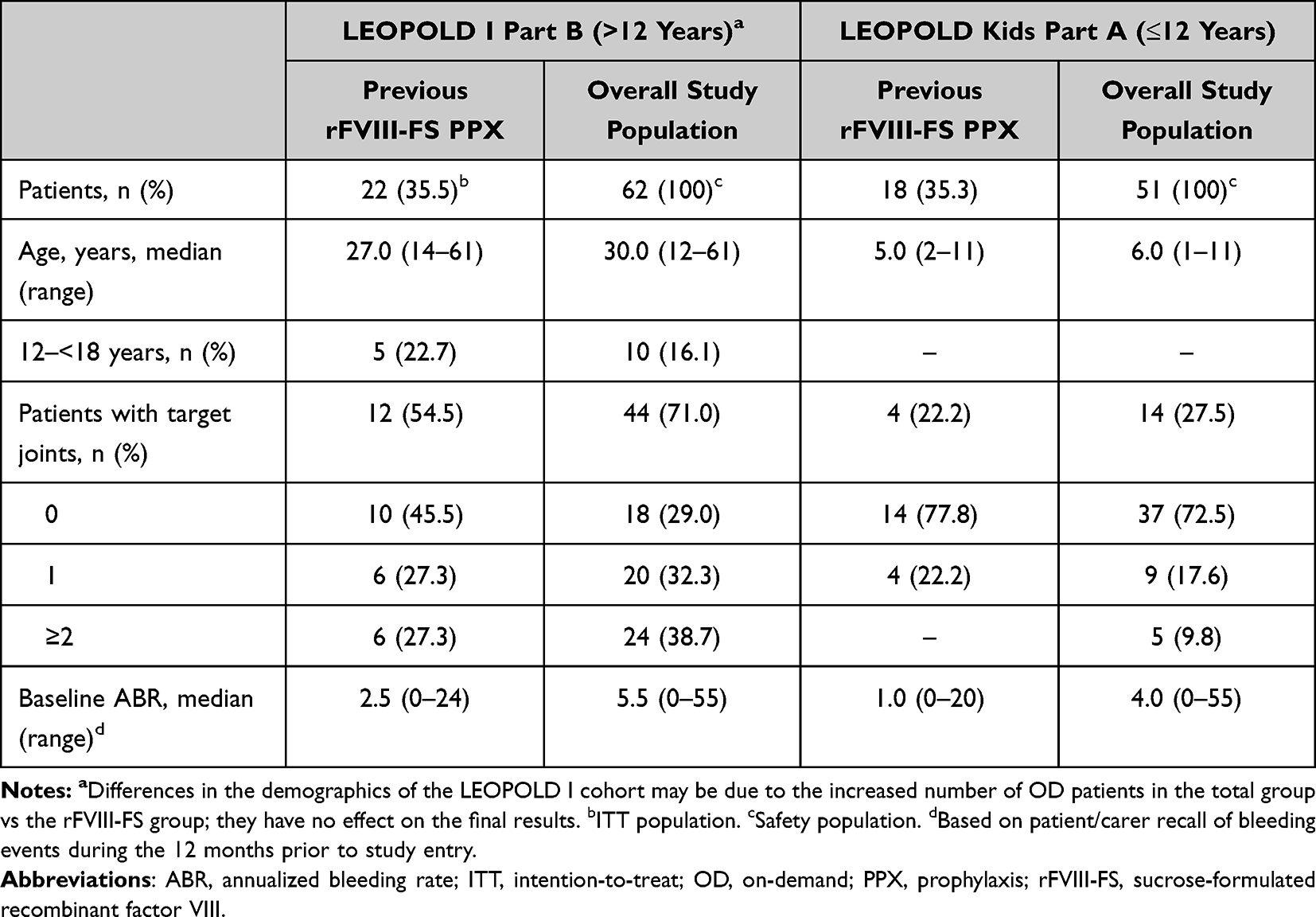 |
Table 1 Baseline Demographics and Disease Characteristics |
The majority of patients maintained the same dosing frequency compared with rFVIII-FS in both the LEOPOLD trials (Figure 2). Most patients were receiving prophylaxis three times weekly during the pre-study period and following switching from rFVIII-FS to octocog alfa. The majority of patients in the LEOPOLD I Part A trial switched to a higher dose of octocog alfa compared with rFVIII-FS, at their physician’s discretion. The median (range) prescribed dose of previous rFVIII-FS prophylaxis treatment was 28.4 (10.0–44.9) IU/kg. The corresponding value for octocog alfa prophylaxis was 30 (20.0–40.0) IU/kg.
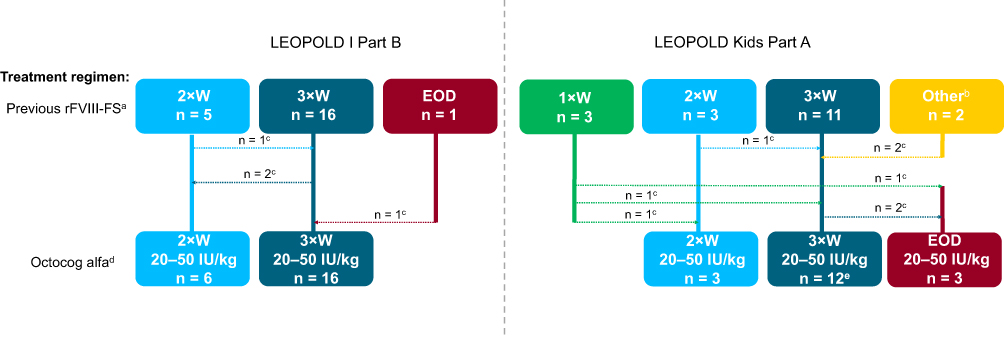 |
Figure 2 Treatment regimen switching from rFVIII-FS to octocog alfa. aData unavailable for previous rFVIII-FS dose; recommended dose for rFVIII-FS prophylaxis is 25 IU/kg 3×W (adults) or 25 IU/kg EOD (children). bThese two patients had an estimated mean dosing frequency prior to the study of 0.39 doses per week and 4.02 doses per week, respectively. cDotted lines represent the overall shift in prophylaxis regimen between pre-study and the start of the respective LEOPOLD studies; bold lines indicate the overall number of patients in each regimen at the start of each LEOPOLD study in relation to the respective previous rFVIII-FS regimens. dPatients were assigned to respective octocog alfa dosing regimens at the investigator’s discretion; the majority of patients remained on their previous dosing interval. eOne patient was represented in more than one previous treatment category. Abbreviations: 1×W, once weekly; 2×W, twice weekly; 3×W, three times weekly; EOD, every other day; rFVIII-FS, sucrose-formulated recombinant factor VIII. |
Bleeding Outcomes
Of patients who switched from rFVIII-FS prophylaxis, 31.8% and 55.6% of prophylaxis patients had zero bleeds in LEOPOLD I Part B and LEOPOLD Kids Part A, respectively. Zero bleed data prior to entry to LEOPOLD I and LEOPOLD Kids studies were not available. In LEOPOLD I Part B (n = 22), median (quartile, Q1; Q3) total ABR decreased from 2.5 (0.0; 9.0) in the 12 months of rFVIII-FS treatment prior to study entry, to 1.0 (0.0; 6.8) (Figure 3) – a reduction in total median ABR of 60%. In LEOPOLD I Part B, median (Q1; Q3) trauma-related ABR was 0.0 (0.0; 1.0).
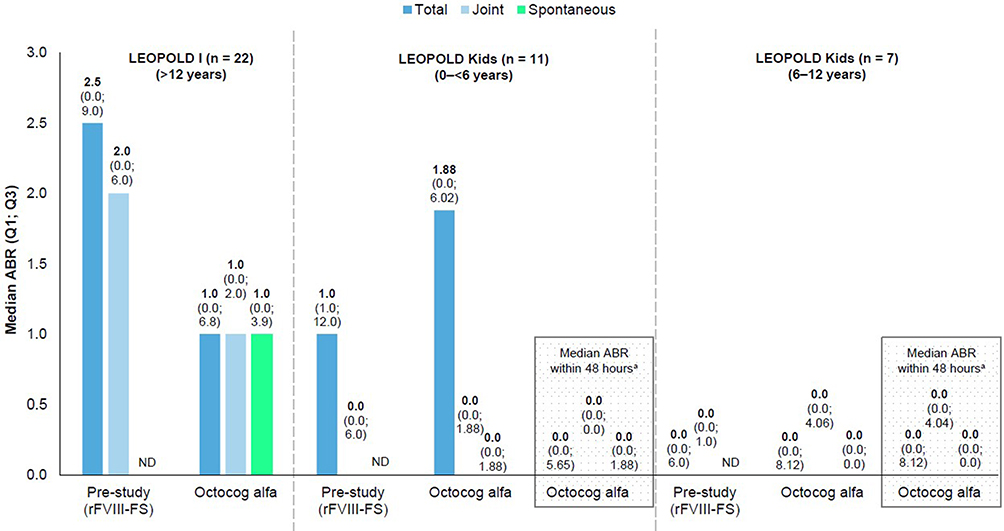 |
Figure 3 ABR outcomes by age in patients previously treated with rFVIII-FS who switched to octocog alfa prophylaxis in the LEOPOLD studies. aThe primary efficacy endpoint in the LEOPOLD Kids study was ABR occurring within 48 hours after prophylaxis infusion, selected because of variable treatment intervals in children, for whom a low infusion frequency could also be related to venous access problems. Abbreviations: ABR, annualized bleeding rate; ND, no data; Q, quartile; rFVIII-FS, sucrose-formulated recombinant factor VIII. |
In LEOPOLD Kids Part A (n = 18), median (Q1; Q3) total ABR decreased from 1.0 (0.0; 6.0) in the 12 months of rFVIII-FS treatment prior to study entry, to 0.0 (0.0; 6.02); median (Q1; Q3) total ABR within 48 hours after octocog alfa prophylaxis was 0.0 (0.0; 5.65). Trauma-related median (Q1; Q3) ABR for all patients (n = 18) was 0.0 (0.0; 2.0). In the 0–<6 years cohort, median (Q1; Q3) total ABR in the pre-study was 1.0 (1.0; 12.0), whereas upon switching to octocog alfa, median (Q1; Q3) total ABR within 48 hours after prophylaxis was 0.0 (0.0; 5.65) (Figure 3). Trauma-related median (Q1; Q3) ABR in this cohort was 0.0 (0.0; 5.65), whereas this was zero within 48 hours after prophylaxis. In the 6–12 years cohort, median total ABRs in the pre-study and the LEOPOLD Kids Part A study were zero; trauma-related median ABR was also zero in the study. The median total ABR within 48 hours after prophylaxis in this cohort was also zero. Median joint ABRs in the pre-study and the LEOPOLD Kids Part A study were zero for both 0–<6 and 6–12 years cohorts. Joint median ABRs within 48 hours after prophylaxis were also zero for both 0–<6 years and 6–12 years cohorts. While pre-study spontaneous ABRs were not available for the 0–<6 and 6–12 years cohorts, spontaneous median ABRs were zero for both cohorts treated with octocog alfa. Similarly, spontaneous median ABRs within 48 hours after prophylaxis were zero for both 0–<6 and 6–12 years cohorts.
Octocog Alfa Utilization
For those who switched from rFVIII-FS to octocog alfa prophylaxis treatment, median (Q1; Q3) FVIII utilization and dose per infusion in LEOPOLD I Part B were 5404.7 (3785.1; 5648.6) IU/kg/year and 35.4 (32.1; 37.2) IU/kg, respectively (Table 2). The corresponding median (Q1; Q3) values for the switch cohort in LEOPOLD Kids Part A were 5810.0 (4359.4; 6358.6) IU/kg/year and 35.2 (28.5; 40.1) IU/kg, respectively (Table 2). rFVIII-FS utilization data prior to entry to LEOPOLD I and LEOPOLD Kids studies were not available.
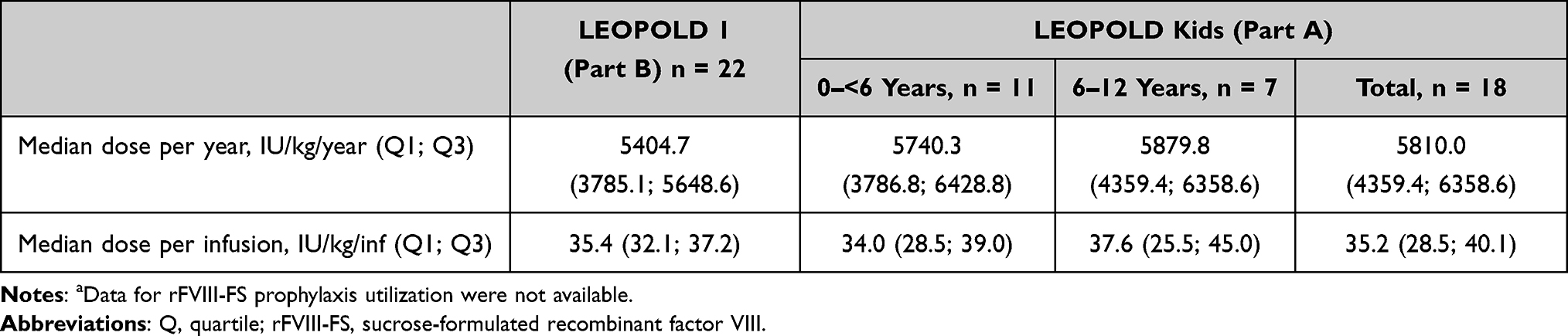 |
Table 2 Octocog Alfa Prophylaxis Utilizationa |
Safety
Overall, the safety profile of those patients who switched from rFVIII-FS to octocog alfa prophylaxis was similar to that of the overall population in the LEOPOLD I Part B and LEOPOLD Kids Part A trials. Of those patients previously treated with rFVIII-FS, AEs were reported in 77.3% (n = 17) and 61.1% (n = 11) of patients in LEOPOLD I Part B and LEOPOLD Kids Part A, respectively (Table 3). No serious or severe AEs were reported (Table 3). There were no discontinuations in LEOPOLD I Part B; one patient discontinued in the LEOPOLD Kids Part A trial (Table 3). No FVIII inhibitors developed in any patients in either study. No deaths or thrombotic events, including thrombotic microangiopathy, were reported.
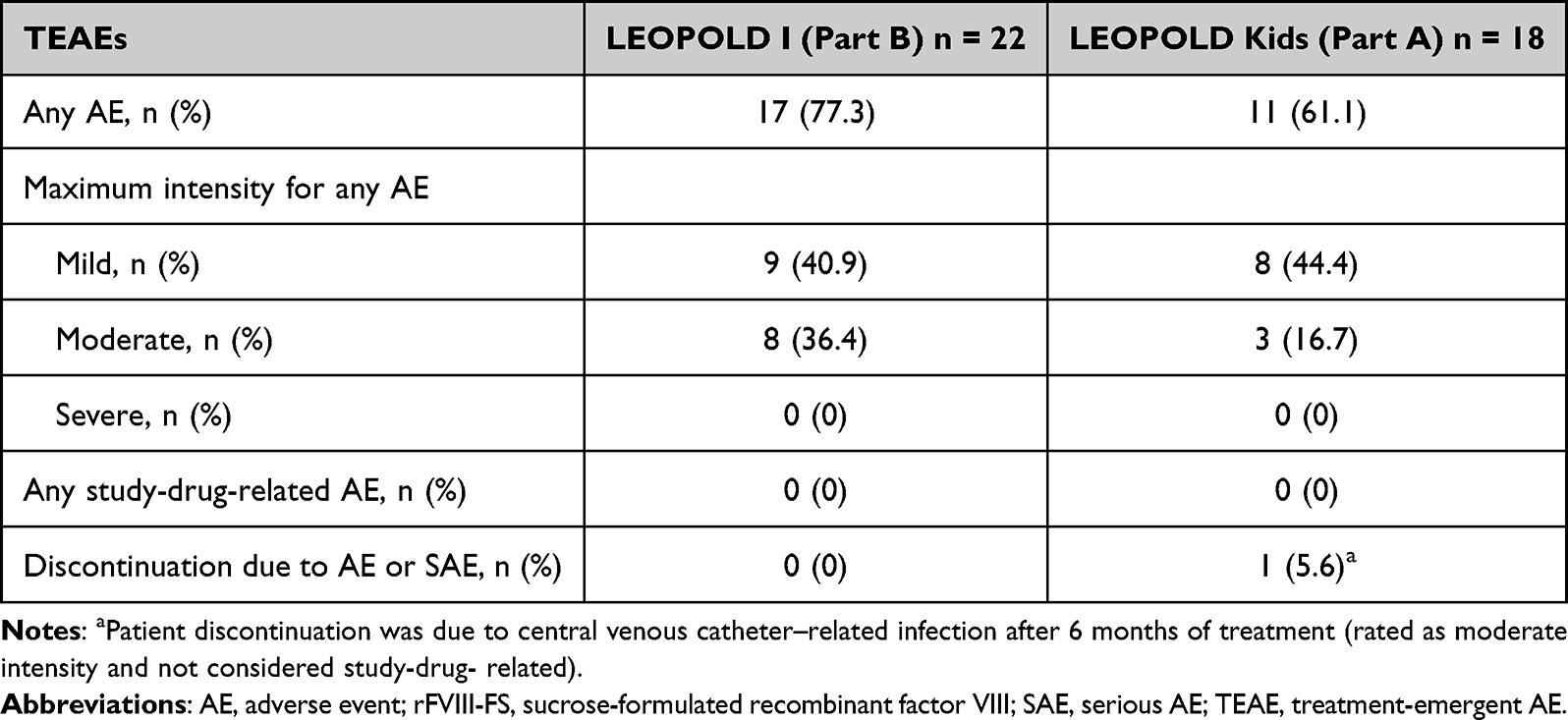 |
Table 3 TEAEs Reported in Patients During the LEOPOLD Studies Previously Treated with rFVIII-FS |
Discussion
The results demonstrate that octocog alfa prophylaxis provides improved bleed protection, defined by a reduction in ABR, compared with similar dosing regimens of rFVIII-FS prophylaxis.
In LEOPOLD I Part B and LEOPOLD Kids Part A, patients who switched from rFVIII-FS to octocog alfa had reduced total ABRs compared with pre-study. In the LEOPOLD Kids Part A study, an increased total ABR was observed in the 0–<6 years cohort when these patients switched from rFVIII-FS to octocog alfa. This observed increase in ABR could be due to carer recall bias on pre-study bleeding events, leading to a lower or less accurate pre-study ABR. It is also plausible that increased trauma-related complications observed in younger children compared with older children and adults could have resulted in the observed increase in total ABR.19–21 Of note, some patients in the 0–<6 years cohort would have been in their more active developmental stage (eg, toddlers) during the study and hence incurred more injuries compared with their relatively less active developmental stage in the pre-study.20,22 Nevertheless, a reduction in total ABR within 48 hours after prophylaxis (primary efficacy outcome in LEOPOLD Kids Part A) was observed in this cohort after switching to octocog alfa, compared with pre-study. Whilst the primary efficacy endpoint in both LEOPOLD I Part B and LEOPOLD Kids Part A was ABR, this was reported differently. The 48-hour endpoint was selected in LEOPOLD Kids Part A to account for the variable treatment intervals in children, for whom a low infusion frequency could also be related to venous access problems. This post hoc subgroup analysis did not aim to compare results between the LEOPOLD studies.
Joint ABRs were lower in the LEOPOLD I Part B switch cohort, compared with pre-study joint ABRs. Although spontaneous ABRs in LEOPOLD I Part B, and joint and spontaneous ABRs in LEOPOLD Kids Part A were low, a comparison of efficacy is not possible due to unavailability of corresponding pre-study data for these patients. The efficacy results of this subgroup analysis complement those of the LEOPOLD I and LEOPOLD Kids primary analyses.11,12
Most patients in either switch cohort maintained their dosing frequency while switching from rFVIII-FS to octocog alfa in the LEOPOLD trials. Better bleed protection with octocog alfa treatment, compared with rFVIII-FS, was also attained using dosing within the approved recommended range for adults and children (20–40 IU/kg and 25–50 IU/kg, respectively), as evidenced from the median dose per infusion received by the switch cohorts in both studies (35.4 IU/kg in LEOPOLD I Part B and 35.2 IU/kg in LEOPOLD Kids Part A). The overall improved efficacy outcomes with octocog alfa observed in this subgroup analysis indicate that most patients currently receiving rFVIII-FS could switch to octocog alfa for better bleed protection. As supported by the dosing recommendations, these data suggest that octocog alfa can also be used with fewer infusions compared with rFVIII-FS.4,5
Octocog alfa was well tolerated in the switch cohorts, with a safety profile similar to that of the overall population in both LEOPOLD studies.11,12 There were no study-drug-related AEs or serious AEs in either of the switch cohorts. No patients discontinued treatment in LEOPOLD I Part B; one patient in LEOPOLD Kids Part A discontinued treatment due to non-study-drug-related central venous catheter-related infection. No immunogenicity concerns were observed, with no patients developing FVIII inhibitors. Switching from rFVIII-FS to octocog alfa prophylaxis did not result in FVIII inhibitor development — a major concern among physicians and patients who are reluctant in switching from their current FVIII therapy.2,3 The most recent guidelines from the World Federation of Hemophilia state that current evidence indicates no increased risk of inhibitor development upon product switching.1 The better efficacy profile observed with octocog alfa treatment over rFVIII-FS in the LEOPOLD switch cohorts could potentially be attributed to the favorable PK profile of octocog alfa as demonstrated in the LEOPOLD PK assessments.11,15
This analysis has a few limitations. First, this is a post hoc study on a limited number of patients with no intra-individual analysis performed. Second, the pre-study data were collected retrospectively based on patient-reported data on bleeds. Pre-study data collection on bleeds relied on a patient’s recall of bleeding events, with data unavailable for a comparison of joint and/or spontaneous ABRs. However, the LEOPOLD studies were prospective in nature, with data on bleeds while on octocog alfa treatment being noted in patient diaries. Third, some of the patients in the switch cohorts were receiving rFVIII-FS below the recommended dose of 25 IU/kg.4 As this was a global study, in some countries, patients may not have been able to dose according to the prescribing information and thus the data may not be representative of all countries.4 Hence, these patients may have had insufficient prophylaxis prior to study entry. Fourth, the recommended dose of 25 IU/kg for rFVIII-FS is less than the recommended dose for octocog alfa (20–40 IU/kg). Thus, using a higher dose of octocog alfa on the same dosing regimen as rFVIII-FS could also improve ABR. Consequently, the extent of differences observed in ABRs in these patients could be an overestimation. Finally, it is plausible that the observed improvement in ABR could be due to a potential improvement in adherence to treatment plan associated with clinical trials.23 Nevertheless, based on this post hoc analysis and previous PK comparisons, treatment with octocog alfa prophylaxis appears to have a favorable risk–benefit profile compared with rFVIII-FS. TAURUS, a recent observational study investigating real-world prophylaxis, found that patients switching from previous FVIII treatment to octocog alfa had stable annualised bleeding rates, treatment satisfaction and adherence over a period of 1–2 years with no new or unexpected safety concerns; indicating that for patients with moderate-to-severe haemophilia A, octocog alpha remains a useful treatment option.24
Conclusion
The results of this post hoc subgroup analysis indicate that octocog alfa prophylaxis could be an effective and improved, alternative strategy for individualized treatment for children, adolescent and adult patients with severe hemophilia A, currently on rFVIII-FS treatment.
Abbreviations
ABR, annualized bleeding rate; AE, adverse event; ED, exposure day; FVIII, factor VIII; PK, pharmacokinetic; rFVIII-FS, sucrose-formulated recombinant FVIII.
Full List of Approving IRBs (TUN)
De Videnskabsetiske Komitéer for Region Midtjylland (35001),
Medizinische Einrichtungen der Universität Bonn Ethikkommission an der Medizinischen Fakultät (10001),
Chaim Sheba Medical Center Helsinki Committee Tel-Hashomer (39001),
Comitato Etico a.o. Pugliese-Ciaccio (22002),
Comitato Etico Per Le Attivita’ Biomediche Carlo Romano Universita’ Studi Federico II (22004),
Comitato Etico Per La Sperimentazione Clinica Della Provincia Di Vicenza (22003),
Comitato Di Etica Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico (22001),
Komisja Bioetyczna przy Inst. Hematologii i Transfuzjogii (18001, 18002),
University of Pretoria, Prinshof Campus (37002),
University of Witwatersrand WITS Human Research Ethics Committee (Medical) (37001),
Ciutat Sanitària i Universitaria de la Vall d’Hebron (24001),
Comité Étic d’Investigació Clínica, Complejo Hospitalario Universitario A Coruña
Servicio Galego de Saude – SERGAS Comité Ético de Investigación Clínico de Galicia (24002),
Hospital Central de Asturias Secretaría del Comité Ético de Investigación Clínica (24006),
Hospital Universitari i Politècnic La Fe Comité Etico de Investigación Clínica / Fundación para la investigación (24003),
Regionala Etikprövningsnämnden i Göteborg (34003),
Ege Universitesi Tip FakultesiEge Universitesi Tip Fakultesi Izmir 1 nolu Klinik Arastirmalar Etik Kurulu (47001, 47002, 47003),
East Midlands - Derby Ethics Committee,
Children’s Hospital Boston Committee on Clinical Investigation (14002),
Children’s Mercy Hospital Pediatric Institutional Review Board (14009),
St. Joseph’s Hospital Tampa Florida (14001),
University Hospitals Case Medical Center IRB for Human Investigation (14005),
University of California – Davis IRB Administration (14006).
Data Sharing Statement
Availability of the data underlying this publication will be determined later according to Bayer’s commitment to the EFPIA/PhRMA “Principles for responsible clinical trial data sharing”. This pertains to scope, time point and process of data access. As such, Bayer commits to sharing upon request from qualified scientific and medical researchers, patient-level clinical trial data, study-level clinical trial data, and protocols from clinical trials in patients for medicines and indications approved in the United States (US) and European Union (EU) as necessary for conducting legitimate research. This applies to data on new medicines and indications that have been approved by the EU and US regulatory agencies on or after January 01, 2014. Interested researchers can use www.clinicalstudydatarequest.com to request access to anonymized patient-level data and supporting documents from clinical studies to conduct further research that can help advance medical science or improve patient care. Information on the Bayer criteria for listing studies and other relevant information is provided in the Study sponsors section of the portal. Data access will be granted to anonymized patient-level data, protocols and clinical study reports after approval by an independent scientific review panel. Bayer is not involved in the decisions made by the independent review panel. Bayer will take all necessary measures to ensure that patient privacy is safeguarded.
Acknowledgments
The authors thank Graeme Baldwin of Darwin Healthcare Communications (Oxford, England) for providing medical writing support, which was fully funded by Bayer, in accordance with Good Publication Practice (GPP3) guidelines.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study and the LEOPOLD studies were funded by Bayer.
Disclosure
G. Kenet: is a member of the Board of Directors/advisory committees and has received consultancy fees from ASC therapeutics, Roche, Sobi, Uniquore, Bayer, Pfizer, BioMarin, Takeda, Roche, Novo Nordisk, Sanofi, PI Healthcare and CSL Behring; has received research funding from BSF, Opko Biologics, Bayer, Pfizer, Roche, Alnylam (Sanofi) and Shire; has received honoraria for participation in speakers’ bureaus from Bayer, BPL, CSL, Roche, Sanofi-Genzyme, Sobi, Spark, Uniquore, Pfizer, Takeda, BioMarin and Novo Nordisk. T. Moulton: is a Bayer employee. B. M. Wicklund: has received consultancy fees from Bayer, Genentech, Novo Nordisk and Shire. S. P. Ahuja: has received consultancy fees from Genentech, Sanofi Genzyme, CSL Behring and XaTek, Inc.; has received patent royalties and research funding from XaTek, Inc. He also has a patent with royalties for ClotChip and a patent for TraumaChek issued to US Patent office. M. Escobar is a member of the advisory boards and/or has received consultancy fees from Biomarin, Novo Nordisk, CSL Behring, Genentech/Roche, Sanofi, Takeda, Pfizer, Bayer, Kedrion, Hemobiologics/LFB, National Hemophilia Foundation, UniQure. J. Mahlangu is a member of the scientific advisory committee of Amgen, Bayer, Baxalta, Biogen, Biotest, CSL Behring, Catalyst Biosciences, Novo Nordisk, Roche and Spark; has received research grants from Bayer, Biogen, BioMarin, CSL Behring, Novartis, Sanofi, Spark, Takeda, Novo Nordisk, Sobi, Roche, Uniqure; has received honoraria for participation in speakers’ bureaus from Alnylam, Bayer, Biogen, Biotest, ISTH, Novo Nordisk, Pfizer, Roche, Sobi, Shire, and World Federation of Hemophilia. The authors report no other conflicts of interest in this work.
References
1. Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26(Suppl 6):1–158. doi:10.1111/hae.14046
2. Santagostino E, Auerswald G, Benson G, et al. Switching treatments in haemophilia: is there a risk of inhibitor development? Eur J Haematol. 2015;94(4):284–289. doi:10.1111/ejh.12433
3. Iorio A, Puccetti P, Makris M. Clotting factor concentrate switching and inhibitor development in hemophilia A. Blood. 2012;120(4):720–727. doi:10.1182/blood-2012-03-378927
4. Kogenate-w-vial-adapter prescribing information. Bayer; 2019.
5. Kovaltry. prescribing information. Bayer; 2016.
6. Kreuz W, Gill JC, Rothschild C, et al. Full-length sucrose-formulated recombinant factor VIII for treatment of previously untreated or minimally treated young children with severe haemophilia A: results of an international clinical investigation. Thromb Haemost. 2005;93(3):457–467. doi:10.1160/TH03-10-0643
7. Collins P, Faradji A, Morfini M, Enriquez MM, Schwartz L. Efficacy and safety of secondary prophylactic vs. on-demand sucrose-formulated recombinant factor VIII treatment in adults with severe hemophilia A: results from a 13-month crossover study. J Thromb Haemost. 2010;8(1):83–89. doi:10.1111/j.1538-7836.2009.03650.x
8. Abshire TC, Brackmann HH, Scharrer I, et al. Sucrose formulated recombinant human antihemophilic factor VIII is safe and efficacious for treatment of hemophilia A in home therapy--International Kogenate-FS Study Group. Thromb Haemost. 2000;83(6):811–816. doi:10.1055/s-0037-1613925
9. Manco-Johnson MJ, Kempton CL, Reding MT, et al. Randomized, controlled, parallel-group trial of routine prophylaxis vs. on-demand treatment with sucrose-formulated recombinant factor VIII in adults with severe hemophilia A (SPINART). J Thromb Haemost. 2013;11(6):1119–1127. doi:10.1111/jth.12202
10. Kavakli K, Yang R, Rusen L, Beckmann H, Tseneklidou-Stoeter D, Maas Enriquez M. Prophylaxis vs. on-demand treatment with BAY 81-8973, a full-length plasma protein-free recombinant factor VIII product: results from a randomized trial (LEOPOLD II). J Thromb Haemost. 2015;13(3):360–369. doi:10.1111/jth.12828
11. Saxena K, Lalezari S, Oldenburg J, et al. Efficacy and safety of BAY 81-8973, a full-length recombinant factor VIII: results from the LEOPOLD I trial. Haemophilia. 2016;22(5):706–712. doi:10.1111/hae.12952
12. Ljung R, Kenet G, Mancuso ME, et al. BAY 81-8973 safety and efficacy for prophylaxis and treatment of bleeds in previously treated children with severe haemophilia A: results of the LEOPOLD Kids trial. Haemophilia. 2016;22(3):354–360. doi:10.1111/hae.12866
13. Oldenburg J, Windyga J, Hampton K, et al. Safety and efficacy of BAY 81-8973 for surgery in previously treated patients with haemophilia A: results of the LEOPOLD clinical trial programme. Haemophilia. 2016;22(3):349–353. doi:10.1111/hae.12839
14. Mahlangu J, Lopez Fernandez MF, Santagostino E, et al. BAY 81-8973 demonstrated efficacy, safety and joint status improvement in patients with severe haemophilia A in the LEOPOLD I extension for ≤2 years. Eur J Haematol. 2020;104(6):594–601. doi:10.1111/ejh.13402
15. Shah A, Delesen H, Garger S, Lalezari S. Pharmacokinetic properties of BAY 81-8973, a full-length recombinant factor VIII. Haemophilia. 2015;21(6):766–771. doi:10.1111/hae.12691
16. Ishaque A, Thrift J, Murphy JE, Konstantinov K. Over-expression of Hsp70 in BHK-21 cells engineered to produce recombinant factor VIII promotes resistance to apoptosis and enhances secretion. Biotechnol Bioeng. 2007;97(1):144–155. doi:10.1002/bit.21201
17. Garger S, Severs J, Regan L, et al. BAY 81-8973, a full-length recombinant factor VIII: manufacturing processes and product characteristics. Haemophilia. 2017;23(2):e67–e78. doi:10.1111/hae.13148
18. Shah A, Solms A, Garmann D, et al. Improved pharmacokinetics with BAY 81-8973 versus antihemophilic factor (recombinant) plasma/albumin-free method: a randomized pharmacokinetic study in patients with severe hemophilia A. Clin Pharmacokinet. 2017;56(9):1045–1055. doi:10.1007/s40262-016-0492-2
19. Kulkarni R, Presley RJ, Lusher JM, et al. Complications of haemophilia in babies (first two years of life): a report from the centers for disease control and prevention universal data collection system. Haemophilia. 2017;23(2):207–214. doi:10.1111/hae.13081
20. Bradshaw CJ, Bandi AS, Muktar Z, et al. International study of the epidemiology of paediatric trauma: PAPSA research study. World J Surg. 2018;42(6):1885–1894. doi:10.1007/s00268-017-4396-6
21. Barg AA, Livnat T, Budnik I, et al. Emicizumab treatment and monitoring in a paediatric cohort: real-world data. Br J Haematol. 2020;191(2):282–290. doi:10.1111/bjh.16964
22. Chaudhary S, Figueroa J, Shaikh S, et al. Pediatric falls ages 0–4: understanding demographics, mechanisms, and injury severities. Inj Epidemiol. 2018;5(Suppl 1):7. doi:10.1186/s40621-018-0147-x
23. Mancuso ME, Oldenburg J, Boggio L, et al. High adherence to prophylaxis regimens in haemophilia B patients receiving rIX-FP: evidence from clinical trials and real-world practice. Haemophilia. 2020;26(4):637–642. doi:10.1111/hae.14018
24. Santoro C, Fuh B, Le PQ, et al. Efficacy and safety in patients with haemophilia A switching to octocog alfa (BAY 81-8973): final results of the global real-world study, Taurus. Eur J Haematol. 2023;110(1):77–87. doi:10.1111/ejh.13876
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.