")
Back to Journals » Drug Design, Development and Therapy » Volume 17
Synthesis of Amino Acids Bearing Halodifluoromethyl Moieties and Their Application to p53-Derived Peptides Binding to Mdm2/Mdm4
Authors Vaas S , Zimmermann MO , Klett T , Boeckler FM
Received 18 February 2023
Accepted for publication 1 April 2023
Published 25 April 2023 Volume 2023:17 Pages 1247—1274
DOI https://doi.org/10.2147/DDDT.S406703
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Video abstract presented by Frank M Boeckler.
Views: 133
Sebastian Vaas,1 Markus O Zimmermann,1 Theresa Klett,1 Frank M Boeckler1,2
1Department of Pharmacy and Biochemistry, Eberhard Karls Universität Tübingen, Laboratory for Molecular Design and Pharmaceutical Biophysics, Institute of Pharmaceutical Sciences, Tübingen, 72076, Germany; 2Institute for Bioinformatics and Medical Informatics (IBMI), Eberhard Karls Universität Tübingen, Tübingen, 72076, Germany
Correspondence: Frank M Boeckler, Molecular Design and Pharmaceutical Biophysics, Institute of Pharmaceutical Sciences, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 8 (Haus B), Tübingen, D-72076, Germany, Tel +49 7071 29 74567, Fax +49 7071 29 5637, Email [email protected]
Introduction: Therapeutic peptides are a significant class of drugs in the treatment of a wide range of diseases. To enhance their properties, such as stability or binding affinity, they are usually chemically modified. This includes, among other techniques, cyclization of the peptide chain by bridging, modifications to the backbone, and incorporation of unnatural amino acids. One approach previously established, is the use of halogenated aromatic amino acids. In principle, they are thereby enabled to form halogen bonds (XB). In this study, we focus on the -R-CF2X moiety (R = O, NHCO; X = Cl, Br) as an uncommon halogen bond donor. These groups enable more spatial variability in protein–protein interactions. The chosen approach via Fmoc-protected building blocks allows for the incorporation of these modified amino acids in peptides using solid-phase peptide synthesis.
Results and Discussion: Using a competitive fluorescence polarization assay to monitor binding to Mdm4, we demonstrate that a p53-derived peptide with Lys24Nle(ϵNHCOCF2X) exhibits an improved inhibition constant Ki compared to the unmodified peptide. Decreasing Ki values observed with the increasing XB capacity of the halogen atoms (F ≪ Cl < Br) indicates the formation of a halogen bond. By reducing the side chain length of Nle(ϵNHCOCF2X) to Abu(γNHCOCF2X) as control experiments and through quantum mechanical calculations, we suggest that the observed affinity enhancement is related to halogen bond-induced intramolecular stabilization of the α-helical binding mode of the peptide or a direct interaction with His54 in human Mdm4.
Keywords: halogen bonding, solid-phase peptide synthesis, unnatural amino acids, fluorescence polarization assay, oncological target, MdmX
Graphical Abstract:

Introduction
Halogenated amino acids and peptides have been the subject of interest in recent research.1–15 Several reviews address the occurrence of natural halogenated amino acids in microbial organisms, bioactive features, eg, in antimicrobial peptides, or the potential of biomimetic engineering in peptide-based drug development and self-assembly. While a wide range of chlorine-, bromine- and iodine-substituted aromatic amino acids in nature are described, the known aliphatic halogenated amino acids are essentially limited to chlorine derivatives due to reactivity of the halogens in aliphatic compounds. In addition, numerous synthetically accessible amino acids have been described in the literature, in particular derivatives of the proteinogenic amino acids phenylalanine, tyrosine, tryptophan, and histidine.16,17
Peptides for therapeutic applications are often derived from natural products or are protein mimetics modified to improve properties such as stability or affinity. These modifications include the use of
Halogenation of amino acid side chains or drugs in general appears to be an effective tool for modulating binding selectivity and increasing binding affinity.21 This modification affects also physico-chemical properties of peptides, such as catabolic stability, lipophilicity, and membrane permeability.22 In addition, chlorine, bromine, and iodine are able to engage in halogen bonding (XB). A halogen bond is a non-covalent bond based on attractive interactions between an electron donor (B) and an electron-deficient region of the halogen (X), the σ-hole, which results from the anisotropic electron distribution of the halogen.23 This interaction is strongly directed along the C–X∙∙∙B axis, with the best interaction energies found at an angle of 180° with a deviation of up to 30° and an X∙∙∙B distance of about 3–4 Å.24,25 Electron donors in proteins are typically the backbone carbonyl of the main chain25–28 and oxygen or nitrogen atoms of electron-rich side chains (eg, aspartate, glutamate),29 serine, histidine,30 or sulfur in methionine.31
As described, halogens bound to aromatic systems are particularly stable relative to aliphatic compounds. In this work, instead of this conventional approach, we establish amino acids with CF2Br ethers and CF2X acetamides (X = Cl, Br), where two electron-withdrawing fluorine atoms are used to stabilize the C–X bond. So far, amino acid-derived compounds with sp3-hybridized CF2Br moiety have been described mainly as precursor for the synthesis of difluoromethylene building blocks.32,33 Applications of such moieties to fragment-based drug discovery, their interactions in small molecule crystals and computational studies with respect to their use in molecular design are reported separately (Vaas S, Zimmermann MO, Boeckler FM. Personal communication, March, 2023).
First, we performed bottom-up synthesis of Fmoc-protected phenylalanine with a bromodifluoromethylether function in either meta or para position. In addition, we prepared the halodifluoromethylamides by one-step syntheses from Fmoc-protected lysine and 2,4-diaminobutyric acid. Fmoc-protection of the amine was necessary for its use in solid-phase peptide synthesis (SPPS). For evaluation, the synthesized amino acids were incorporated into 12-mer p53-derived peptides by automated SPPS.
The tumor suppressor protein p53 is an important oncologic target and is known to be implicated in most of all human cancers.34,35 Rescuing p53 mutations and targeting the p53 regulatory network has been a focal point of our research.24,36–43 The proteins Mdm2 and Mdm4 are negative regulators of p53, and their interaction is a common model system for evaluating inhibition of protein–protein interactions (PPIs).44 Both proteins bind to the same sequence in the intrinsically disordered N-terminus of p53 with their structurally very similar N-terminal domains.45,46 The surface region of the binding site of Mdm2 and Mdm4 are small hydrophobic clefts47,48 with limited molecular interaction capabilities. The critical amino acids Phe19, Trp23 and Leu26 (p53 numbering) involved in the binding are found in almost every peptide sequence described in literature.49 Mutations of these amino acids lead to a drastic loss of binding affinity.50 These properties hinder the substitution of these three amino acids by other proteinogenic and non-proteinogenic amino acids and complicate the introduction of new PPI contacts that could lead to stronger interactions. Therefore, in our study, we consider the enhancement of direct peptide–protein interactions, as well as the stabilization of the helix formation of the p53 peptide by forming an intramolecular interaction.
We used this PPI model to evaluate our p53-derived sequences containing the CF2X-bearing amino acids by determination of Ki values using a competitive fluorescence polarization (FP) assay51 with an N-terminal fluorescein-labeled high-affinity peptide as probe.38,49,52
Materials and Methods
Peptide Synthesis
All peptides were synthesized with free N-terminus and amidated C-terminus on a 50 μmol scale by SPPS using the Fmoc strategy. The automated SPPS was carried out on a MultiSynTech Syro I peptide synthesizer (Biotage, Uppsala, Sweden). After deprotection and cleavage from the Rink amide resin (TentaGel® HL RAM, particle size: 75 µm, capacity: 0.38 mmol g–1, Rapp Polymere, Tübingen, Germany) with cleavage solution (85% (v/v) trifluoroacetic acid, 5.5% (wt) phenol, 4.5% (v/v) triisopropylsilane, 5% (v/v) H2O), the resin was washed three times with DCM and the resulting mixture was concentrated under reduced pressure. Subsequently, the peptides were precipitated twice in cold diethyl ether (–20 °C). The crude peptides were purified by a reversed-phase HPLC system (PuriFlash 4250, Interchim, Montluçon, France) equipped with a ReproSil-XR 120 C18 column (5 μm, 120 Å, 30×250 mm, Dr. Maisch GmbH, Ammerbuch-Entringen, Germany) and a linear gradient elution with a mobile phase composed of eluent A (99.9% (v/v) H2O and 0.1% (v/v) trifluoroacetic acid) and eluent B (80% (v/v) acetonitrile, 19.9% (v/v) H2O, and 0.1% (v/v) trifluoroacetic acid) at a flow rate of 30 mL min–1. The collected fractions were freeze-dried. The molecular masses were determined by HR-ESI mass spectrometry (Bruker maXis 4G, Bruker Corporation, Billerica, MA, USA) The purity of the peptides was assessed by analytical HPLC on an UltiMate 3000 HPLC system (Thermo Fisher Scientific Inc. Waltham, MA, USA) equipped with a ReproSil-XR 120 C18 column (4.6 × 150 mm, 5 µm, 120 Å, Dr. Maisch GmbH, Ammerbuch-Entringen, Germany). The purity of all synthesized test peptides was >95% as determined by HPLC analysis.
Protein Expression and Purification
Mdm2 (2–125): Expression and purification were mainly adopted from the literature and modified.53 The pminiRSET/pRESTa/pGEX-2T_GST_MDM2 construct was transformed into E. coli Rosetta 2 or BL 21 (DE3) pLysS cells (Novagen, Merck, Darmstadt, Germany) and incubated at 37 °C overnight on LB medium plates with ampicillin (50 μg mL–1) and chloramphenicol (37 μg mL–1). Cells were transferred with LB medium and the same antibiotics into a 9-fold volume of the same media and grown to an OD600 = 1.0 at 37 °C and 180 rpm as a pre-culture. Twenty mL of this culture was used to inoculate 4 L of 2xYT containing the antibiotics. These main cultures were grown at 37 °C and 220 rpm until an OD600 = 0.4, then the temperature was reduced to 25 °C and IPTG was added to final concentration of 0.5 mM. The expression lasted for 16 h. Cultures were centrifuged at 4 °C, 4000 rpm for 30 min in a J6-MI (Beckman-Coulter, Brea, CA, USA) centrifuge. The pellet was resuspended with ice-cold lysis buffer (50 mM TRIS, 500 mM NaCl, 5 mM DTT, 1 mM EDTA, 0.1% (v/v) Triton X-100, 50 μM PMSF, pH of 8.0). After adding DNase and RNase, the suspension was lysed by sonication. The suspension was centrifuged at 4 °C and 18,500 rpm for 1 h in an Avanti J-30-I (Beckman-Coulter, Brea, CA, USA). The supernatant was sterile filtered (0.22 μM filter, rapid-FILTERMAX, TTP®, Trasadingen, Switzerland) and loaded onto a GST-affinity column (GE XK 1, Amintra Glutathione Resin, Expedeon, Heidelberg, Germany) equilibrated with elution buffer (50 mM TRIS, 500 mM NaCl, 5 mM DTT, 1 mM EDTA, pH of 8.0). The column was washed with 5 CV of elution buffer. The protein was eluted with elution buffer containing 10 mM reduced glutathione using a linear gradient elution from 0% elution buffer to 100% in 15 min. The fractions were pooled and thrombin-digested at pH 8.0 and 4 °C (250 units high active thrombin per 40 mL) overnight. After protein concentration using a Vivaspin® Turbo 15 (MWCO = 5 kDa, Sartorius, Göttingen, Germany), the protein was loaded onto a size-exclusion gel filtration column (HiLoad 26/60 Superdex 75 prep grade, GE Healthcare, Chicago, IL, USA) equilibrated with SEC buffer (5 mM TRIS, 50 mM NaCl, 5 mM β-ME, pH of 8.0). The fractions collected were concentrated to approximately 60 µM, then 10% (v/v) glycerol was added, the mixture was flash-frozen in liquid nitrogen and aliquots were stored at –80 °C. The purity of the protein was monitored by SDS-PAGE after each purification step. The protein mass was confirmed by ESI-MS and the protein concentration was calculated via UV-spectroscopy using the molar extinction coefficient 10,430 M–1 cm–1 at 280 nM from the ProtParam program on the EXPASY server.
Mdm4 (16–116, C17S): Expression and purification were mainly adopted from the literature and modified.38,54 The pET24a(+)_HLT_MDM4 construct was transformed into E. coli Rosetta 2 or BL 21 (DE3) pLysS cells (Novagen, Merck, Darmstadt, Germany) and incubated at 37 °C overnight on LB medium plates with kanamycin (50 μg mL–1) and chloramphenicol (37 μg mL–1). Cells were transferred with LB medium and the same antibiotics into a 9-fold volume of the same media and grown to an OD600 = 1.0 at 37 °C and 180 rpm as a pre-culture. Ten mL of this culture was used to inoculate 6 L of 2xYT containing the antibiotics. These main cultures were grown at 37 °C and 220 rpm until an OD600 = 0.6, then the temperature was reduced to 20 °C and IPTG was added to final concentration of 1.0 mM. The expression lasted for 16 h. Cultures were centrifuged at 4 °C, 4000 rpm for 30 min in a J6-MI (Beckman-Coulter, Brea, CA, USA) centrifuge. The pellet was resuspended with ice-cold lysis buffer (50 mM sodium phosphate, 300 mM NaCl, 10 mM imidazole, 5 mM β-ME, pH of 8.0). After adding DNase and RNase, the suspension was lysed by sonication. The suspension was centrifuged at 4 °C and 18,500 rpm for 1 h in an Avanti J-30-I (Beckman-Coulter, Brea, CA, USA). The supernatant was sterile filtered (0.22 μM filter, rapid-FILTERMAX, TTP®, Trasadingen, Switzerland) and loaded onto a Nickel-NTA column (HisTrap FF, GE Healthcare, Chicago, IL, USA) equilibrated with lysis buffer. The column was washed with 5 CV of lysis buffer. The Protein was eluted with elution buffer (50 mM sodium phosphate, 300 mM NaCl, 250 mM imidazole, 5 mM β-ME, pH of 8.0) using a linear gradient elution from 0% elution buffer to 100% in 15 min. His-tag were cleaved with 1 mg mL–1 TEV protease per 5 mL protein sample while dialyzing with regenerated cellulose sleeve (ZelluTrans/Roth, Dialysiermembran T1, MWCO = 3500 Da, Carl Roth, Karlsruhe, Germany) in 5 L lysis buffer without imidazole overnight on stirrer at 4 °C. The His-tag and His-tagged TEV were removed using a reverse nickel column run. After protein concentration using a Vivaspin® Turbo 15 (MWCO= 5 kDa, Sartorius, Göttingen, Germany), the protein was loaded onto a size-exclusion gel filtration column (HiLoad 26/60 Superdex 75 prep grade, GE Healthcare, Chicago, IL, USA) equilibrated with SEC buffer (25 mM sodium phosphate, 150 mM KCl, 5 mM β-ME, pH of 7.2). The collected fractions were concentrated to approximately 50 µM, then 10% (v/v) glycerol was added, the mixture was flash-frozen in liquid nitrogen and aliquots were stored at –80 °C. The purity of the protein was monitored by SDS-PAGE after each purification step. The protein mass was confirmed by ESI-MS and the protein concentration was calculated via UV-spectroscopy using the molar extinction coefficient 7450 M–1 cm–1 at 280 nM from the ProtParam program on the EXPASY server.
Fluorescence Polarization Assay
Peptide stock solutions (800 µM) of fluorescence-labeled probe and test peptides were prepared in DMSO and stored at –20 °C. The buffer of a thawed Mdm2 or Mdm4 protein solution was exchanged using a desalting column (HiPrepTM 26/10 Desalting, Cytiva, Marlborough, MA, USA). FP assay buffer (pH 7.2 at 25 °C) contained 25 mM potassium phosphate, 150 mM ionic strength (NaCl), 5 mM DTT. The protein was concentrated with Vivaspin® Turbo 4 or 15 (MWCO = 5 kDa, Sartorius, Göttingen, Germany) at 4 °C and 4000 rpm where appropriate. BSA (0.2 mg mL–1) was added just before the FP measurement. All FP experiments were performed using a CLARIOstar plate reader (emission filter: 530 nM, excitation filter: 482 nM, and dichroic mirror: 504 nM. BMG Labtech, Ortenberg, Germany) with black non-binding polystyrene 96-well microplates (GBO, Frickenhausen, Germany).
Kd values of the fluorescence probe (FAM-LTFEHYWAQLTS-CONH2) against Mdm2 and Mdm4 were determined by direct titration. 11-step serial dilutions of protein were prepared in FP assay buffer. To obtain the final protein concentration with 5% (v/v) DMSO, 190 µL of the dilutions were mixed in a 96-well plate with 5 µL DMSO and 5 µL of 800 nM fluorescence-labeled probe (final concentration: 20 nM) in DMSO. A twelfth measurement point contained only DMSO instead of probe. Each protein concentration was measured as quadruplicate. After 30 min incubation at 25 °C, measurements were taken three times at 10 min intervals and the results were averaged. This series of measurements was repeated at least three times to determine reliable Kd values and protein concentrations in a range of 50–70% of detected FP signal for competitive FP assay. It is recommended to re-determine these values for each protein batch. For the competitive FP assay, 11-step serial dilutions of test peptide were prepared in DMSO. To obtain the final test peptide concentration with 5% (v/v) DMSO, 5 µL of the dilutions were mixed in a 96-well plate with 5 µL of 800 nM fluorescence-labeled probe (final concentration: 20 nM) in DMSO and 190 µL protein solution (protein concentration determined during direct titration experiment) in FP assay buffer. A twelfth measurement point contained only DMSO instead of test peptide. For experiments with 10% (v/v) DMSO instead of 5% (v/v) DMSO, the protein solution was reduced to 180 µL and additional 10 µL DMSO was added. Each test peptide concentration of the competitive assay was measured in quadruplicates and each series of measurements was repeated at least three times analogous to direct titration.
All normalized fluorescence polarization measurements were fitted with four parameter logistic regression (4PL) in OriginPro 2020 (OriginLab, Northampton, MA, USA) using the following equation,

where A1 is the minimum value and A2 is the maximum value that can be obtained, x0 is the point of inflection and p is the Hill’s slope of the curve. The inhibition constant Ki was determined using the following equation,

where [I]50 denotes the concentration of the free inhibitor at 50% inhibition, [L]50 is the concentration of the free labeled ligand at 50% inhibition, [P]50 is the concentration of the free protein at 0% inhibition, and Kd is the dissociation constant of the protein-ligand complex. [I]50, [L]50 and [P]50 were determined as described in literature.51
Circular Dichroism Spectroscopy
CD measurements were performed on a Jasco J-720 (Jasco, Easton, MD, USA) at 20 °C using a quartz flow cell with a 2 mm path length. Peptides were dissolved in PBS (pH 7.4) at a concentration of 150 µM. Spectra were recorded at 50 nm min–1 with a bandwidth of 2 nm and averaged over 2–10 scans. The PBS baseline was subtracted from each spectrum. The mean residue weight ellipticity [θ] was calculated using the following equation,
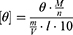
where θ is the measured ellipticity in [mdeg], M is the molar mass in [g mol–1], n is the number of amino acid residues in the peptide, m is the dissolved mass in [g], V is the volume of the solvent in [cm3], and l is the path length in [cm].
Computational Methods
PDB Database Screening
Peptides bound to HIV-1 protease, Mdm2, and Mdm4 were analyzed for putative new halogen bonds. For this, aromatic carbon positions of peptide amino acids were decorated with CF2Br or OCF2Br groups. The attached substituents were rotated along all rotatable bonds in steps of 5°. Severe clashes with the surroundings were filtered out. Hits with a putative halogen bond angle of >160° and an interaction distance of <4 Å were saved as PSE files. This was done using a custom PyMOL/Python script. The data was then sighted manually.
Generation of Conformers
The side chain of Lys24 in chain P (starting at the Cα atom) in the crystal structure of 2Z5T was modified into the bromodifluoroacetylated variant and protonated using the Protonate 3D feature of Molecular Operating Environment (MOE) 2018.0101 at default settings. Then, a conformational search of this modified side chain was conducted using the Conformational Search feature of MOE at default settings, resulting in 162 conformers.
Binding Site Representation
In two crystal structures (PDB IDs: 2Z5T and 3DAB), the surroundings of Lys24 were carefully selected and used as an interaction site for the modified lysine. The interaction site consists of:
Peptide residues in 2Z5T and 3DAB: Phe19 (C, O, Cα), Ser20 (full), Asp21 (full), Leu22 (side chain removed), Trp23 (full), Lys24 (full), Leu25 (N, Cα).
Protein residues in 2Z5T: MetT50 (C, O, Cα), His51 (full), Tyr52 (side chain removed), Leu53 (side chain removed), Gly54 (full), Gln55 (full), Tyr56 (side chain removed), Ile57 (side chain removed), Met58 (full), Val59 (N, Cα).
Protein residues in 3DAB: Met53 (C, O, Cα), His54 (full), Tyr55 (side chain removed), Leu56 (side chain removed), Gly57 (full), Gln58 (full), Tyr59 (side chain removed), Ile60 (side chain removed), Met61 (full), Val62 (N, Cα).
Next, the interaction site representation was protonated using MOE and the Protonate 3D feature at default settings.
Generation of Interaction Site Variants Using the Conformers
Using a custom Python script, the interaction site representations were altered using the previously generated conformers of the modified lysine side chain as follows: The positions of the Cα and Cβ atoms of Lys24 were used to match the modified conformers, thus replacing the natural side chain. For each conformer, 12 variations of the interaction site were generated by rotating the modified side chain around the Cα and Cβ bond in steps of 30°. Next, generated interaction site variants resulting in severe clashes between the modified lysine side chain and the remaining peptide residues or the protein were removed. This resulted in 450 interaction site variants for 2Z5T and 420 for 3DAB.
Quantum Mechanical Calculations
Optimization of the remaining interaction site variants was performed using TPSS-D3/def-SV(P)55–58 within TURBOMOLE 7.4.1.59 Heavy atoms of the protein and peptide backbone were kept frozen during optimization. The convergence criterion was set to 10−6. Due to the high atom number (115 heavy atoms, 108 hydrogen atoms), $scfdamp was altered to: start=5.000 step=0.050 min=0.500. Multipole accelerated RI-J was activated ($marij). For 2Z5T, 247 converged structures were obtained and for 3DAB 304 structures.
Results and Discussion
PDB Database Screening and Sequence Selection
Trying to rationalize possible applications of CF2X-bearing amino acid side chains in PPI, we scanned HIV-1 protease, Mdm2, and Mdm4 Protein Data Bank (PDB) entries via aromatic C atom decoration of phenylalanine and tyrosine using a PyMOL/Python script. The attached substituents were rotated along its individual axes in five-degree steps. Hits with suitable C–X∙∙∙B angle and X∙∙∙B distance (B = N, O) were checked. Incidentally, the search was limited to the entries of resolved crystal structures deposited in the PDB.
Two selected findings are shown in Figure 1. Figure 1A shows a peptide phenylalanine decorated with a CF2Br unit in para position in a CA-p2 substrate/HIV-1 protease complex (PDB ID: 1A8K; peptide sequence: RVL-r-FEA-Ahx-NH2),60 with a σ-hole angle of 165.8° and a distance of 3.4 Å to the Pro81 backbone oxygen. Another decoration result was found in a PMI (N8A mutant)/Mdm2 complex (PDB ID: 3LNZ; peptide sequence: TSFAEYWALLSP),50 as shown in Figure 1B, where a peptide tyrosine was decorated in meta position. The CF2Br ether moiety intramolecularly targets a peptide threonine with a C–Br∙∙∙O angle of 177.2° and an X∙∙∙O distance of 3.0 Å. Suitable peptide decorations with direct interaction between peptide and Mdm2/Mdm4 were not found in contrast to intramolecular side chain interactions, which may be due to the nature of the binding pocket described in the introduction. Inspired by these results, the syntheses of halogenated Fmoc-protected phenylalanines were planned and performed as described. Due to chemical stability considerations, the synthesis of amino acids with a catechol substructure (as shown in Figure 1B) was omitted. In addition to the ether function, we also attempted to prepare a thioether and sulfone isostere.
 |
Figure 1 PDB decoration scan of aromatic amino acids in peptides. (A) CF2Br methyl unit attached to a phenylalanine in para position targeting the backbone oxygen of Pro81 in a CA-p2 substrate/HIV-1 protease crystal structure (PDB ID: 1A8K); (B) CF2Br ether moiety attached to a tyrosine in meta position intramolecularly targeting a peptide threonine side chain in a 12-mer peptide inhibitor PMI (N8A mutant)/Mdm2 crystal structure (PDB ID: 3LNZ). |
As a second approach, we synthesized non-aromatic amino acids by acetylation of aminated alkyl side chains and their incorporation into the 17-28p53 wild-type sequence (ETFSDLWKLLPE) mutating Lys24Nle(εNHCOR) and Lys24Abu(γNHCOR) (with R = CH3, CF2H, CF3, CF2Cl and CF2Br). Due to the large number of degrees of freedom in the lysine side chain, computational decoration scans of PDB structures were not considered appropriate here. Besides the importance of being located in the sequence binding to Mdm2/Mdm4, the p53 sequence interacts with mitochondrial E3 ubiquitin protein ligase 1 (MUL1) resulting in ubiquitination of cytoplasmic p53 at Lys24 and subsequent proteasomal degradation.61 Acetylated lysine is no longer expected to cross-link interchain with the C-terminal glycine of ubiquitin. Known mutation Lys24Arg in p53 abolishes ubiquitination by MUL1 and the somatic mutation Lys24Asn is implicated in sporadic cancer.62 Therefore, we were interested in evaluating the p53 sequence by modifying the existing Lys24 via different substituted acetyl groups.
Amino Acid Synthesis
Synthesis of Phenylalanine Derivatives
The amino acid precursors 16–18 containing CF2Br functionality were synthesized from methylbenzene derivatives as shown in Scheme 1. A brominatable methyl group was mandatory to introduce the diethylacetamidomalonate function, which was subsequently converted to the amino acid function.
 |
Scheme 1 Synthesis of 3-/4-methylbenzene derivatives containing the CF2Br moiety as precursors for amino acid synthesis. Reagents and conditions: (a) Ethyl bromodifluoroacetate, act. Cu0, argon atmosphere, 60 °C, 26 h; (b) 1 M aq. K2CO3/MeOH (1:1), rt, 18 h; (c) (1) (COCl)2, DMF (cat.), DCM, rt, 3 h; (2) DMAP, sodium-N-hydroxy-2-thiopyridone, BrCCl3, argon atmosphere, 300 W sunlamp, rt, 20 h; (d) (1) NaH, 1,4-dioxane, argon atmosphere, rt, 30 min; (2) potassium 2-bromo-2,2-difluoroacetate (1), 1,4-dioxane, argon atmosphere, 80 °C, 20 h; (e) (1) (COCl)2, DMF (cat.), DCM, rt, 3 h; (2) DMAP, sodium-N-hydroxy-2-thiopyridone, BrCCl3, argon atmosphere, 120 °C, 2 h; (f) (1) NaH, DMF, rt, 0 °C; (2) CF2Br2, DMF, 0 °C, 3 h; (g-1) NBS, ABCN, CCl4 or benzene, argon atmosphere, reflux, 8 h; (g-2) diethylacetamidomalonate, K2CO3, KI, ACN, argon atmosphere, reflux, 18 h; (h) mCPBA, DCM, rt, 18 h. |
Ethyl 2,2-difluoro-2-(p-tolyl)acetate (2) was synthesized by means of Ullmann coupling using hydrochloric acid activated copper(0) as catalyst.63,64 The obtained ester was hydrolyzed under basic conditions to halogenate the free carboxylic acid (3) by Barton decarboxylation. The Barton decarboxylation requires the in situ generation of a Barton ester using acid chloride generated with oxalyl chloride and sodium-N-hydroxy-2-thiopyridone as reagent,65 which is followed by a photoinduced radical decarboxylation. With a suitable radical trapping agent such as BrCCl3, the introduction of a bromine is possible, as performed in our case to obtain 6 with a yield of 41%.
p-Cresol (28) and m-Cresol (29) were elaborated to give bromodifluoromethoxylated derivatives using a reaction sequence with analogous intermediates.66 The free carboxylic acids 4 and 5 were generated via nucleophilic substitution directly with the reagent potassium bromodifluoroacetate (1) under basic conditions bypassing ester hydrolysis. In contrast to photolyzed Barton decarboxylation at methyl groups, this reaction with methoxy groups requires higher temperatures around 80°C for 2 hours and provided slightly higher yields with 51% for 7 and 54% for 8.
The CF2Br thioether moiety could be directly obtained from the corresponding thiol (30) in a one-step synthesis, due to the higher nucleophilicity of thiolates compared to alcoholates. In the first step, sodium hydride was used to generate the thiolate, which was reacted with reagent CF2Br2 to obtain 9 with a yield of 63%.66,67 The synthesized compounds with CF2Br moieties (10–13) were all brominated at the methyl position and, without further purification, converted to the amino acid precursors 14–17.68 The yield of this two-step synthesis varied from 45% for 14 to 77% for 17. In addition, 17 was oxidized with mCPBA to obtain the sulfone derivative 18 with a yield of 73%.67
Synthesizing amino acid functionality while maintaining the CF2Br moiety was a particular challenge. Two possible routes to obtain Fmoc-protected amino acids are shown in Scheme 2. Route (a) presents a one-pot-synthesis under heat with harsh acidic conditions leading to ester hydrolysis, amide hydrolysis and decarboxylation, followed by subsequent Fmoc-protection of amine functionality and chiral column chromatography. The simplest reaction condition is refluxing in concentrated hydrochloric acid. This and different modulations of temperature, reaction time, acids, acid concentrations, organic solvents and organic solvent contents did not result in the desired products.
 |
Scheme 2 Two possible routes to obtain Fmoc-protected amino acids. Route (a) presents acidic conditions and route (b) alkaline reaction conditions. |
With bases such as NaOH, it was possible to generate amino acids according to route (b) while retaining the acetamido and CF2Br moiety (Scheme 2). This was achieved for 15 and 16, while 14, 17 and 18 also degraded with this method (Scheme 3). In principle, this method can be carried out in one step under heat, see 19 with a yield of 57%. Alternatively, hydrolysis can be done first at room temperature under basic condition and decarboxylation in a second step without base under heat.
 |
Scheme 3 Synthesis of Fmoc-protected amino acid derivatives containing CF2Br moiety. Reagents and conditions: (a-1/2) 15, NaOH, MeOH/ H2O (3:1), rt, 1 h, then 80 °C, 3 h; or: (a-1) 16, NaOH, MeOH/ H2O (3:1), rt, 4 h; (a-2) THF/ H2O (3:1), 3 h, 70 °C; (b) 19, 0.1 M phosphate buffer (pH 8), CoCl2, Amano Acylase, rt, 6 h; or: 20, 1 M aq. NaOH, pH 7.0–10, Amano Acylase, rt, 4 h; (c) Fmoc-Cl, Na2CO3, 1,4-dioxane, rt, 4 h. |
The second method resulted in a higher yield of 93% for the conversion of 16 to 20. An additional advantage of this synthetic route (b) was that an enzymatic resolution of enantiomers was performed in the following step, which eliminated the need for separation via chiral column chromatography. Amano Acylase was used for acetamide cleavage of only the
Non-Aromatic Amino Acids with Halodifluoro Acetyl Amide Moiety
Another method to obtain Fmoc-protected amino acids with a CF2X moiety is to modify side chains with amine function by coupling with derivatives of acetic anhydride or other acetic acid-based reagents to their corresponding CF2X acetamides. The advantage is that the desired products can be achieved in a one-step synthesis due to the commercial availability of the reagents. Fmoc-
For the studies presented in this work, acetylated derivatives of Fmoc-protected
 |
Scheme 4 Synthesis of Fmoc-protected |
 |
Table 1 Comparison Between Yields of Fmoc-Protected Nle and Abu Derivatives |
Acetylation of
Peptide Synthesis
Chromatographic reversed-phase purification did not reveal any significant difference in the purity and yield of the peptides containing the CF2Br ether and CF2X amide moieties compared to the peptides prepared entirely from standard Fmoc-protected amino acids (Tables 2 and S1). Consequently, the modified amino acids can be exposed to strongly basic as well as strongly acidic conditions without any loss of the CF2X group or other peptide degradation. Even mild reducing agents such as TIPS, which acts as a hydride donor scavenger, do not appear to have any adverse effects.69
 |
Table 2 Synthesized Peptide Sequences with X as Modified Amino Acid |
Fluorescence Polarization Assay
FP Assay – CF2Br Ether
For the competitive assay, a protein working concentration of 30 nM was determined for Mdm2 and 60 nM for Mdm4 by direct titration using 20 nM high-affinity probe, and the Kd values determined were 14.3 nM and 38.2 nM, respectively.
All tested peptides TPE-1 to TPE-3 bound to Mdm2 and Mdm4 (Figure S1 and Table 3). Initially, a DMSO concentration of 5% (v/v) was used. Very similar values were obtained with Ki values ranging from 199 nM to 258 nM for Mdm2 and slightly better for Mdm4 with 71 nM to 82 nM. Due to the very moderate solubility of the peptides, the measurements were repeated in 10% (v/v) DMSO. As a result, all peptides displaced the probe more effectively, but again, no significant differences between peptides were observed in terms of inhibitory properties. The measured Ki values range from 171 nM to 189 nM for Mdm2 and 55 nM to 65 nM for Mdm4. Only at Mdm2 TPE-1 displaced the probe better than TPE-2 (5% (v/v) DMSO: 217 nM vs 258 nM, and 10% (v/v) DMSO: 171 nM vs 189 nM). Figure S1 notably illustrates by the nearly identical shape of the curve that all measured differences are not significant.
 |
Table 3 Summary of Competitive Fluorescence Polarization Assay Results of Measured IC50 and Calculated Ki Values for TPE-1, TPE-2, TPE-3 with 20 nM Fluorescence-Labeled Probe and 30 nM Mdm2 (Kd = 14.6 nM) or 60 nM Mdm4 (Kd = 38.2 nM), Measured in 5% or 10% DMSO. Values are Shown as Mean ±SD (Each Measuring Point as Quadruplicate; Number of Measurement Series: n=3) |
As shown in Figure 1B, we hypothesize that the putative XB interaction occurs on the solvent exposed side of the peptide. This may have a detrimental effect on peptide affinity. Experience with the solubilities of the modifications investigated had shown that these decrease in aqueous environments in the following order: CH3 > CF3 > CF2Br. Accordingly, the CF2Br moiety was disadvantageous compared to the other two groups. Furthermore, the solubilizing hydroxy functionality contained in the original tyrosine was removed.
FP Assay – CF2Br Amide
Here, we focus on the more decisive 17-28p53 peptide and its acetylated derivatives. For the competitive assay with the 17-28p53-derived peptides, a Mdm4 working concentration of 30 nM was determined by direct titration using 20 nM high-affinity probe, and the probe Kd determined was 11.5 nM.
Initially, peptides 17-28p53 and TPA-2 to TPA-6 with modified Lys24Nle(εNHCOR) (R = CH3, CF2H, CF3, CF2Cl, CF2Br) were measured by FP-displacement assay with Mdm4. As shown in Figure 2A and Table 4, significant inhibitory differences were observed. A Ki value of 307 nM was measured for the wild-type sequence of 17-28p53. In relation to this, acetylated lysine (TPA-2) had a 1.66-fold higher Ki value at 509 nM, and difluoro acetylated TPA-3 had a 1.15-fold higher Ki value at 353 nM. For TPA-4 (CF3), TPA-5 (CF2Cl), and TPA-6 (CF2Br), we observed an increase in inhibitory properties, as we would expect from an XB interaction. TPA-4 had a Ki of 202 nM, TPA-5 a Ki of 112 nM and TPA-6 a Ki of 82 nM. All peptides displaced the probe better than the 17-28p53 peptide, the chlorine derivative by 3-fold and the bromine derivative by 4-fold. The difference was quite remarkable, considering that of the 215 atoms/111 heavy atoms (110 heavy atoms in case of TPA-3) that constitute each of the peptides TPA-3 to TPA-6, only one atom was exchanged for another atom.
 |
Table 4 Summary of Competitive Fluorescence Polarization Assay Results of Measured IC50 and Calculated Ki Values for TPA-1 to TPA-12 with 20 nM Fluorescence-Labeled Probe and 30 nM Mdm4 (Kd = 11.5 nM) in 5% DMSO. Values are Shown as Mean ±SD (Each Measuring Point as Quadruplicate; Number of Measurement Series: n=3) |
 |
Figure 2 Relative fluorescence polarization in [%] of competitive fluorescence polarization assay for TPA-1 (17−28p53) and TPA-2 to TPA-12 with 20 nM fluorescence-labeled probe and 30 nM Mdm4 (Kd = 11.5 nM) in 5% DMSO. Measurement series are shown as mean ±SD (each measuring point as quadruplicate; number of measurement series: n=3). (A) TPA-1 (17−28p53) with Lys24 and TPA-2 to TPA-6 with Nle24 derivatives. (B) TPA-7 with Dab24 and TPA-8 to TPA-12 with Abu24 derivatives. (C) normalized Ki values of TPA-1 to TPA-12 relative to TPA-1 (17−28p53). |
However, it was not yet possible to ascertain from the trend of the Ki whether a halogen bond was involved. To understand the binding motif more clearly, peptides were tested which contained Lys24Dab and Lys24Abu(γNHCOR) derivatives with only two methylene units in the side chain instead of four methylene units like Lys/Nle derivatives. The measurements of corresponding peptides TPA-7 to TPA-12 are shown in Figure 2B. All peptides displaced the probe with Ki values of 423 nM to 647nM worse than the 17-28p53 peptide, as listed in Table 4. From this, we deduced that the side chain length was relevant for the interaction of the halogenated acetyl groups of TPA-4, TPA-5, and TPA-6, which led them to show better inhibition than 17-28p53. Figure 2C shows the normalized Ki values of all tested peptides relative to TPA-1 (17-28p53).
We have attempted to approach a plausible binding motif hypothesis in silico via known crystal structures deposited in the PDB, as we were not able to obtain a crystal structure of the best binding peptide TPA-6 with Mdm4. Only a few appropriate Mdm4/p53-peptide crystal structures were available. Deposited Mdm4 crystal structures in complex with the p53 peptide were 3DAB and 3DAC (both human),48 or structurally highly similar 2Z5S and 2Z5T (both danio rerio).70 The crystal cells contain up to 4 Mdm4/p53-peptide pairs in which not all Lys24 of the peptide are fully resolved. In 2Z5T (chain F), Lys24 in association with Ser20 and Asp21 forms a complex with a water molecule (Figure 3A).
 |
Figure 3 Mdm4/15−29p53 crystal structure (chain E and chain F of PDB ID: 2Z5T).70 (A) a water molecule is coordinated by the side chains of Ser20, Asp21, and Lys24. (B) illustration of modified Lys24Nle(εNHCOCF2Br) in 15−29p53 peptide. |
As shown in Figure 3B, we modified Lys24 of the p53 peptide in 2Z5T and in 3DAB, by attaching a CF2Br acetamide group. In a next step, we carried out a conformational analysis of the modified side chain to sufficiently cover the conformational space. After sorting out conformers involving clashes with the surrounding crystal structure, we performed a geometry optimization using TPSS-D3/def-SV(P).55–58 During optimization, heavy atoms of the peptide and protein backbone were kept frozen. Out of the 247 converged structures of 2Z5T, 36 structures yielded a halogen bond with a distance <4 Å and an interaction angle >160° (Figure 4 and Table S2). Only one of these halogen bonds already existed before the optimization process. In the case of 3DAB, 304 structures converged, with 54 halogen bonds using the thresholds as above (Figure 4 and Table S3). Further analysis of the data revealed motifs with additional interactions, such as 33 H∙∙∙F contacts of the fluorine atoms (<3.0 Å, >150°), 25 hydrogen bonds (HBs, <3.5 Å, >150°) of the amide O, and five HBs of the amide NH (<3.5 Å, >150°, Table S4) for 2Z5T as well as 50 interactions of fluorine atoms and three amide NH contacts in the case of 3DAB (Table S5). In some cases, combinations of these interactions can occur in the same structure. For 2Z5T, we found in 84 of the total 247 structures at least one intermolecular or intramolecular interaction from the CF2Br acetamide group toward Mdm4 or the peptide itself, respectively. For 3DAB, 100 of the total 304 structures exhibited such positive results (intramolecular or intermolecular interactions). The remaining 163 (2Z5T)/204 (3DAB) converged structures mostly started at a great distance from potential interaction partners and consequently found none during optimization. In summary, 49% of the 184 positive results involve an XB, 39% involve contact with at least one of the two fluorine atoms, 11% involve an HB with the amide oxygen, and 4% involve an HB with the amide NH.
 |
Figure 4 Statistical representation of the conformational analysis and subsequent geometry optimization process. (A) schematic representation of all XB interactions found between the bromine atom and XB acceptors in 15−29p53 peptide and in Mdm4 protein. (B) number of matches of the respective XB acceptors. (C) dependencies of the XB (C–Br∙∙∙N/O) angle [°] to the XB (Br∙∙∙N/O) distance [Å]. |
We split the XBs into two groups as highlighted by color in Figure 4A: 23 (2Z5T)/41 (3DAB) intramolecular XBs were found between the CF2Br and different XB acceptors in the peptide and 13 (2Z5T)/13 (3DAB) XBs targeting the protein. Within the peptide, the bromine found six appropriate XB acceptors and five in Mdm4. Interestingly, the XB acceptor Asp21 side chain in p53 stand out with 35 matches and the histidine side chain [His51 in zebrafish Mdm4 (2Z5T) and His54 in human Mdm4 (3DAB)] in Mdm4 with 18 matches. Replacing the coordinated water molecule, the intramolecular XB aims at the potentially negatively charged carboxylic acid of Asp21 and thereby would conceivably stabilize helix formation (Figure 5A and C). Additional data from CD spectra (Figure S2) indicate that no increased helicity was observed in solution and that this possible intramolecular binding would only be present if the peptide, which is intrinsically disordered in solution, adopts a helical structure in the binding site of Mdm4. As shown in Figure 5B and D, the second proposed interaction possibility leading to affinity enhancement is an XB with the unprotonated Nδ of the His51/54 side chain in Mdm4. In both proposals, the XB acceptor also aligns to the XB donor during optimization process (Figure 5). In contrast to the intramolecular contact, the interactions to Mdm4 have a more concerted character, which is due to the longer distance to be bridged. Both described interactions are not or only with difficulty feasible using the shorter amino acid derivatives with Lys24Abu(γNHCOR). Since the distance to Mdm4 was very long (>9 Å from Cα) a direct interaction with His51/54 of Mdm4 was rather excluded, but an intramolecular contact with Asp21 is conceivable (>7 Å from Cα). Less frequently found XB interactions, which were labelled (a), (b), (c), (e), (f), (g), (h), (i), (j), and (l) in Figure 4, are depicted in Figure S3 for comparison. In addition, all amino acids within 12 Å distance around Lys24 of the 15-29p53 peptide chain are shown as a reference for the putative interaction space in Figure S4 for zebrafish Mdm4 (2Z5T) and human Mdm4 (3DAB).
 |
Figure 5 Final conformations after geometry optimization with Lys24Nle(εNHCOCF2Br) in modified crystal structures of PDB IDs 2Z5T and 3DAB.48,70 Mdm4 protein in beige. 15−29p53 peptide in light blue. (A) 2Z5T: Ten XB matches (d) targeting intramolecularly the Asp21 side chain of 15−29p53 peptide. (B) 2Z5T: Ten XB matches (k) targeting the His51 side chain of Mdm4 protein. (C) 3DAB: 25 XB matches (d) targeting intramolecularly the Asp21 side chain of 15−29p53 peptide. (D) 3DAB: Eight XB matches (k) targeting the His54 side chain of Mdm4 protein. |
Conclusion
In summary, we have established two approaches to synthesize Fmoc-protected amino acids with a CF2X moiety for application in SPPS. First, bottom-up synthesis of phenylalanine-derived 23 and 24 with a CF2Br ether moiety in para or meta position was conducted. Then, we synthesized amino acids with an alkyl side chain (25 and 26) of variable length ((CH2)n, n =1,3) containing a terminal CF2X acetamide moiety (X = Cl, Br). Compared to the 7-step synthesis of ethers, the preparation of the acetamide derivatives from commercially available amines is considerably simpler with only one synthesis step and provides rapid generation of a wide range of building blocks for the SPPS.
The incorporation of Lys24Nle(εNHCOCF2X) into 17-28p53-derived peptides shows decreasing Ki values in the competitive FP-assay towards Mdm4 in the order X = F > Cl > Br, indicating an XB interaction. This trend is not observed in measurements with shorter side chain Lys24Abu(γNHCOCF2X) derivatives. The Lys24Nle(εNHCOCF2Br) derivative shows a 4-fold stronger inhibition constant compared to the unacetylated amine of Lys24. Compared to the ether moiety, the amide increases the solubility significantly and enables the formation of hydrogen bonds as HB donor and HB acceptor in addition to its XB donor functionality. CD spectra and further in silico experiments using published protein crystal structures indicate an intramolecular interaction occurring in the Mdm4-bound, helical state of the peptide. Herein, we found an XB interaction with the carboxylic acid group of Asp21. Likewise, a direct intermolecular XB between the unprotonated Nδ in His54 of human Mdm4 and the CF2Cl or CF2Br moiety of the peptides could explain the increase in affinity.
Future studies will focus on the design of further amino acids bearing CF2X moieties, the identification of high-affinity peptide sequences containing these amino acids, and the investigation of their mode-of-action regarding XB.
Chemistry
General
All chemicals and reagents were obtained from commercial sources and used as received. Reactions were monitored by thin-layer chromatography (TLC) carried out on Merck Kieselgel 60 F254 plates (Merck, Darmstadt, Germany) and visualized under UV light (254 nm and 366 nm) or by analytical high-performance liquid chromatography (HPLC). HPLC was performed on an UltiMate 3000 HPLC system (Thermo Fisher Scientific Inc. Waltham, MA, USA) equipped with a ReproSil-XR 120 C18 column (4.6 × 150 mm, 5 µm, 120 Å, Dr. Maisch GmbH, Tübingen, Germany).
Chromatographic purifications were carried out on an Interchim PuriFlash 4250 or PuriFlash XS520Plus system (Interchim, Montluçon, France) using normal-phase column chromatography (Silica gel 60, particle size: 0.025–0.04 mm and 0.04–0.063 mm, Machery-Nagel, Düren, Germany) or equipped with a C18 column (puriFlash® C18-HP 15µm) using reversed-phase column chromatography. Absorption was detected at 218 nm, 254 nm, 280 nm, and 360 nm.
Mass spectrometry (MS) was performed on AmaZon SL (Bruker Corporation, Billerica, MA, USA) using positive or negative electrospray ionization (ESI) or MSD 5977 (Agilent Technologies Inc., Santa Clara, CA, USA) using electron ionization (EI) and high-resolution mass spectrometry was recorded on a maXis 4G (Bruker Corporation, Billerica, MA, USA) using positive electrospray ionization (HR-ESI-TOF) coupled with an UltiMate 3000 HPLC system (Thermo Fisher Scientific Inc. Waltham, MA, USA).
NMR spectra were acquired with Bruker Avance III HDX 400 spectrometer and Bruker Avance III HDX 700 spectrometers (Bruker Corporation, Billerica, MA, USA). 1H, 13C and 19F NMR spectra were reported as chemical shifts (δ) in parts per million (ppm) in relation to tetramethylsilane (TMS) and were calibrated using the residual peak of the used solvent or, in case of 19F NMR spectra, using the reference peak of C6F6 (163.0 ppm) or TFA (76.0 ppm). Coupling constants (J) were reported in units of hertz (Hz). The following abbreviations were used to describe multiplicities: s (singlet), d (doublet), q (quartet), m (multiplet) and bs (broad singlet).
The purity of all synthesized final compounds was >95% as determined by HPLC analysis.
Fmoc-Protected Amino Acid Synthesis
General Procedure A
An appropriate cresol (10 mmol, 1 eq) was dissolved in dry 1,4-dioxane (30 mL) under argon atmosphere, then NaH (60% dispersion in mineral oil) (11 mmol, 1.1 eq) was added. After 30 min stirring at room temperature, 1 (11 mmol, 1.1 eq) was added and the mixture was stirred at 60 °C until no more cresol can be observed with TLC (approximately 5 to 6 h). The reaction mixture was cooled down to room temperature, then Et2O was added and the mixture was extracted three times with sat. NaHCO3. Et2O was added to the combined aq. phases, then the biphasic mixture was acidified with conc. HCl at 0 °C to pH = 1. The mixture was extracted 3 times with Et2O, the combined organic phases were washed with brine and dried over Na2SO4. After removing the solvent, 1,4-dioxane is still present. The purity was determined using 1H NMR and the crude product was used without further purification for the next synthesis step.
General Procedure B
To a solution of an appropriate carboxylic acid (5 mmol, 1 eq) in DCM (10 mL) was added a catalytic amount of DMF (0.5 mmol, 0.1 eq) and oxalyl chloride (7.5 mmol, 2 M in DCM, 1.5 eq) at 0 °C. The reaction mixture was stirred at room temperature for 3 h, then concentrated in vacuo. The crude acyl chloride was added BrCCl3 (15 mL), DMAP (1.25 mmol, 0.25 eq) and sodium-N-hydroxy-2-thiopyridone (6 mmol, 1.2 eq). The reaction mixture was refluxed at 120 °C for 2 h under argon atmosphere, then concentrated in vacuo. The crude product was purified by silica gel column chromatography.
General Procedure C
In an oven-dried two-necked flask, an appropriate methylbenzene (10 mmol, 1 eq) was dissolved in dry benzene or CCl4 (30 mL) under argon atmosphere, then NBS (11 mmol, 1.1 eq) and ABCN (3 mmol, 0.3 eq) were added. The suspension was refluxed for 8 h. Afterwards, the mixture was cooled down to room temperature and the solid succinimide was filtered off. The filtrate was concentrated in vacuo and the crude product was used without purification for the next synthesis step.
General Procedure D
The crude bromomethylbenzene from General Procedure C (10 mmol, 1 eq), diethylacetamidomalonate (10 mmol, 1 eq), K2CO3 (20 mmol, 2 eq), and KI (10 mmol, 1 eq) were dissolved in dry ACN (60 mL) under argon atmosphere. The suspension was refluxed for 18 h. After that, the mixture was cooled down to room temperature and filtered. The solvent was removed in vacuo and the crude product was purified by silica gel column chromatography.
General Procedure E
To a suspension of an appropriate Fmoc-protected amino acid (10 mmol, 1 eq) and NMM (30 mmol, 3 eq) in dry THF (70 mL) were added dropwise the required anhydride (12 mmol, 1.2 eq) at 0 °C under argon atmosphere. The reaction mixture was stirred at 0 °C for 30 min, then additional anhydride was added dropwise at 0 °C until the suspension becomes a clear solution without any precipitate and the resulting red-brown solution was stirred at room temperature for 2 h. The reaction mixture was quenched by addition of MeOH and concentrated under reduced pressure. The residue was dissolved in EtOAc and washed twice with 10% HCl, then with H2O and brine. The solvent was removed in vacuo and the crude product was purified by reversed-phase column chromatography.
Synthesis Procedures
Potassium 2-bromo-2,2-difluoroacetate (1)
Potassium hydroxide (31.4 g, 619 mmol, 1.0 eq) was dissolved in MeOH (500 mL) at 0 °C, then ethyl bromodifluoroacetate (132 g, 650 mmol, 1.05 eq) was added. The mixture was allowed to warm up to room temperature and stirred for 72 h. The solvent was removed in vacuo to provide the title compound 1 (130 g, 98.6%) as a white solid.
ESI-MS (m/z): [M-K]− 173.0/175.0 (1:1); 19F NMR (376 MHz, DMSO-d6): δ –47.77 (s, 2F).
Ethyl 2,2-difluoro-2-(p-tolyl)acetate (2)
To a solution of 1-iodo-4-methylbenzene 27 (1.0 g, 4.59 mmol, 1 eq) and ethyl bromodifluoroacetate (647 µL, 5.05 mmol, 1.58 g cm−3, 1.1 eq) under argon atmosphere was added activated Cu0 powder (874.4 mg, 13.76 mmol, 1 eq). The reaction mixture was stirred at 60 °C for 26 h. The crude mixture was filtered through a pad of diatomaceous earth and washed with Et2O. The organic layer was washed with sat. NH4Cl (2 × 30 mL) and brine (2 × 30 mL), dried over Na2SO4 and the solvent was removed in vacuo. The crude product was purified by silica gel column chromatography (eluent: PE 40/60/Et2O 98:02) to provide the title compound 2 (651.5 mg, 66.3%) as a colorless oil.
Activation of copper powder: Cu0 powder (874.4 mg, 13.76 mmol, <425 µm) was stirred in aq. 6 M HCl (5 mL) for 10 min at room temperature. The solution was decanted, and the procedure was repeated with H2O (5 mL), MeOH (5 mL) and acetone (5 mL). Before use, the Cu0 powder was dried under vacuum for 15 min.
EI-MS (m/z): [M]+ 214.01; 1H NMR (600 MHz, DMSO-d6): δ 7.47 (d, J = 18.2 Hz, 2H), 7.35 (d, J = 7.9 Hz, 2H), 4.29 (q, J = 7.1 Hz, 2H), 2.35 (s, 3H), 1.21 (t, J = 7.9 Hz, 2H); 13C NMR (151 MHz, CDCl3): δ 169.7 (t, J = 36.8 Hz), 142.0, 129.6, 129.1 (t, J = 25.7 Hz), 125.6, 113.3 (t, J = 252.1 Hz), 21.5; 19F NMR (565 MHz, CDCl3): δ –105.13 (s, 2F).
2,2-Difluoro-2-(p-tolyl)acetic acid (3)
To a solution of 2 (5.18 g, 24 mmol, 1 eq) in MeOH (70 mL) was added aq. 1 M K2CO3 (70 mL). The reaction mixture was stirred at room temperature for 18 h. MeOH was removed in vacuo, then the mixture was diluted with Et2O and the biphasic mixture was extracted 3 times with sat. NaHCO3. The combined aq. layers were cooled to 0 °C and Et2O was added. The biphasic mixture was acidified with conc. HCl to pH = 1. After that, the mixture was extracted three times with Et2O, the combined organic phases were washed with sat. brine and dried over Na2SO4. The solvent was removed in vacuo to obtain the title compound 3 (4.37 g, 97.1%) as a white solid.
ESI-MS (m/z): [M-H]− 185.07; 1H NMR (600 MHz, CDCl3): δ 10.80 (s, 1H), 7.51 (d, J = 8.2 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 2.40 (s, 3H); 13C NMR (151 MHz, CDCl3): δ 163.4 (t, J = 35.0 Hz), 141.5, 129.6, 129.1 (t, J = 25.5 Hz), 125.0 (t, J = 5.8 Hz), 113.6 (t, J = 250.4 Hz), 63.3, 20.8, 13.6; 19F NMR (565 MHz, CDCl3): δ –101.52 (s, 2F).
2,2-Difluoro-2-(p-tolyloxy)acetic acid (4)
The title compound was prepared following General Procedure A using p-cresol 28 (500 mg, 4.62 mmol, 1.0 eq), NaH (60% dispersion in mineral oil; 203 mg, 5.09 mmol, 1.1 eq) and 1 (1.08 g, 5.09 mmol, 1.1 eq) in dry 1,4-dioxane (15 mL). The title compound 4 (882 mg, 94.3%) was obtained as a yellow oil.
ESI-MS (m/z): [M-H]− 201.06; 1H NMR (600 MHz, DMSO-d6): δ 7.22 (d, J = 8.3 Hz, 2H), 7.10 (d, J = 8.4 Hz, 2H), 3.56 (1,4-dioxane), 2.29 (s, 3H); 13C NMR (151 MHz, DMSO-d6): δ 160.8 (t, J = 39.6 Hz), 147.0, 135.9, 130.3, 121.2, 114.3 (t, J = 271.2 Hz), 66.4 (1,4-dioxane), 20.3; 19F NMR (565 MHz, DMSO-d6): δ –76.60 (s, 2F).
2,2-Difluoro-2-(m-tolyloxy)acetic acid (5)
The title compound was prepared following General Procedure A using m-cresol 29 (20.0 g, 185 mmol, 1.0 eq), NaH (60% dispersion in mineral oil; 8.13 g, 203 mmol, 1.1 eq) and 1 (43.3 g, 203 mmol, 1.1 eq) in dry 1,4-dioxane (700 mL). The title compound 5 (31.1 g, 83.4%) was obtained as a yellow oil.
ESI-MS (m/z): [M-H]− 200.85; 1H NMR (400 MHz, DMSO-d6): δ 7.31 (t, J = 7.8 Hz, 1H), 7.17–7.08 (m, 1H), 7.05–7.01 (m, 2H), 2.32 (s, 3H); 13C NMR (101 MHz, DMSO-d6): δ 160.7 (t, J = 39.5 Hz), 149.2, 139.9, 129.7, 127.1, 121.7, 118.1, 114.2 (t, J = 271.6 Hz), 20.8; 19F NMR (376 MHz, DMSO-d6): δ –76.08 (s, 2F).
1-(Bromodifluoromethyl)-4-methylbenzene (6)
To a solution of 3 (418.5 mg, 2.25 mmol, 1.0 eq) in DCM (2.8 mL) was added a catalytic amount of DMF (17.3 µL, 0.23 mmol, 0.95 g cm−3, 0.1 eq) and oxalyl chloride (1.70 mL, 3.37 mmol, 2 M in DCM, 1.5 eq) at 0°C. The reaction mixture was stirred at room temperature for 3 h, then concentrated in vacuo. The crude acyl chloride was added degassed BrCCl3 (7 mL), DMAP (68.7 mg, 0.56 mmol, 0.25 eq) and sodium-N-hydroxy-2-thiopyridone (402.2 mg, 2.70 mmol, 1.2 eq). The reaction mixture was stirred under argon atmosphere and illuminated by a 300 W sun lamp for 20 h, then concentrated in vacuo. The crude product was purified by silica gel column chromatography (eluent: PE 40/60/EtOAc 95:5) to provide the title compound 6 (201.9 mg, 40.6%) as a colorless oil.
EI-MS (m/z): [M]+ 218.9/220.9 (1:1); 1H NMR (400 MHz, CDCl3): δ 7.50 (d, J = 8.3 Hz, 2H), 7.25 (d, J = 7.9 Hz, 2H), 2.40 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 141.8, 135.7 (t, J = 23.5 Hz), 129.4, 124.4 (t, J = 5.1 Hz), 118.8 (t, J = 303.5 Hz), 21.5; 19F NMR (376 MHz, CDCl3): δ –42.57 (s, 2F).
1-(Bromodifluoromethoxy)-4-methylbenzene (7)
The title compound was prepared following General Procedure B using 4 (18.7 g, 92.5 mmol, 1.0 eq), DMF (712 µL, 9.25 mmol, 0.95 g cm−3, 0.1 eq) and oxalyl chloride (69.4 mL, 138.7 mmol, 2 M in DCM, 1.5 eq) in DCM (115 mL). After the first reaction step and concentration of the reaction mixture, BrCCl3 (260 mL), DMAP (2.82 g, 23.1 mmol, 0.25 eq) and sodium-N-hydroxy-2-thiopyridone (16.6 g, 111.0 mmol, 1.2 eq) were added for the second reaction step. The crude product was purified by silica gel column chromatography (eluent: n-hexane/EtOAc 97:3) to provide the title compound 7 (11.2 g, 51.1%) as a yellow oil.
EI-MS (m/z): [M]+ 235.9/237.8 (1:1); 1H NMR (400 MHz, CDCl3): δ 7.20 (d, J = 8.3 Hz, 2H), 7.13 (d, J = 8.7 Hz, 2H), 2.37 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 148.9 (t, J = 2.0 Hz), 137.0, 130.3, 121.3, 115.0 (t, J = 308.2 Hz), 21.0; 19F NMR (376 MHz, CDCl3): δ –15.33 (s, 2F).
1-(Bromodifluoromethoxy)-4-methylbenzene (8)
The title compound was prepared following General Procedure B using 5 (30.0 g, 148.4 mmol, 1.0 eq), DMF (2.8 mL, 14.8 mmol, 0.95 g cm−3, 0.1 eq) and oxalyl chloride (111 mL, 222 mmol, 2 M in DCM, 1.5 eq) in DCM (115 mL). After the first reaction step and concentration of the reaction mixture, BrCCl3 (490 mL), DMAP (4.5 g, 37.1 mmol, 0.25 eq) and sodium-N-hydroxy-2-thiopyridone (22.1 g, 148.4 mmol, 1.0 eq) were added for the second reaction step. The crude product was purified by silica gel column chromatography (eluent: n-hexane/EtOAc 99:1) to provide the title compound 8 (19.0 g, 53.9%) as a yellow oil.
EI-MS (m/z): [M]+ 236.0/238.0 (1:1); 1H NMR (400 MHz, CDCl3): δ 7.31–7.26 (m, 1H), 7.14 (d, J = 7.5 Hz, 1H), 7.06 (d, J = 4.8 Hz, 1H), 2.40 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 151.0, 140.3, 129.5, 127.9, 122.1, 118.4, 114.7 (t, J = 308.4 Hz), 21.5; 19F NMR (376 MHz, CDCl3): δ –16.37 (s, 2F).
(Bromodifluoromethyl)(p-tolyl)sulfane (9)
To a solution of 4-methylbenzenethiol 30 (2.0 g, 16.1 mmol, 1.0 eq) in dry DMF (32 mL) was added NaH (60% dispersion in mineral oil, 966 mg, 24.2 mmol, 1.5 eq) at 0 °C. The reaction mixture was stirred at room temperature for 30 min, then CF2Br2 was added at 0 °C and the mixture was stirred further 3 h at 0 °C. The remaining NaH was quenched with H2O (400 mL) and the mixture was extracted with EtOAc (3 × 100 mL). The combined organic layers were washed with brine (100 mL), dried over Na2SO4 and concentrated in vacuo. The crude product was purified by silica gel column chromatography (eluent: PE 40/60/Et2O 99:1) to provide the title compound 9 (2.55 g, 62.5%) as a colorless oil.
EI-MS (m/z): [M]+ 251.9/253.9 (1:1); 1H NMR (400 MHz, CDCl3): δ 7.54 (d, J = 8.1 Hz, 2H), 7.24 (d, J = 7.9 Hz, 2H), 2.40 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 141.8, 136.6, 130.4, 123.9, 119.7 (t, J = 338.3 Hz), 21.6; 19F NMR (376 MHz, CDCl3): δ –22.34 (s, 2F).
1-(Bromodifluoromethyl)-4-(bromomethyl)benzene (10)
The title compound was prepared following General Procedure C using 6 (1.17 g, 5.30 mmol, 1.0 eq), NBS (1.04 g, 5.83 mmol, 1.1 eq) and ABCN (389 mg, 1.59 mmol, 0.3 eq) in CCl4 (15 mL). The crude product 10 was used without further purification for the next synthesis step of 14.
1-(Bromodifluoromethoxy)-4-(bromomethyl)benzene (11)
The title compound was prepared following General Procedure C using 7 (10.6 g, 44.8 mmol, 1.0 eq), NBS (8.76 g, 49.2 mmol, 1.1 eq) and ABCN (3.28 g, 13.4 mmol, 0.3 eq) in CCl4 (125 mL). The crude product 11 was used without further purification for the next synthesis step of 15.
1-(Bromodifluoromethoxy)-3-(bromomethyl)benzene (12)
The title compound was prepared following General Procedure C using 8 (2.00 g, 8.44 mmol, 1.0 eq), NBS (1.65 g, 9.28 mmol, 1.1 eq) and ABCN (619 mg, 2.53 mmol, 0.3 eq) in benzene (25 mL). The crude product 12 was used without further purification for the next synthesis step of 16.
(Bromodifluoromethyl)(4-(bromomethyl)phenyl)sulfane (13)
The title compound was prepared following General Procedure C using 9 (2.44 g, 9.63 mmol, 1.0 eq), NBS (1.88 g, 10.6 mmol, 1.1 eq) and ABCN (706 mg, 2.89 mmol, 0.3 eq) in CCl4 (27 mL). The crude product 13 was used without further purification for the next synthesis step of 17.
Diethyl 2-acetamido-2-(4-(bromodifluoromethyl)benzyl)malonate (14)
The title compound was prepared following General Procedure D using crude 10 (5.30 mmol, 1.0 eq), diethylacetamidomalonate (1.15 g, 5.30 mmol, 1.0 eq), K2CO3 (1.47 g, 10.6 mmol, 2.0 eq) and KI (880 mg, 5.30 mmol, 1.0 eq) in ACN (30 mL). The crude product was purified by silica gel column chromatography (eluent: PE 40/60/EtOAc 75:25) to provide the title compound 14 (1.14 g, 49.1% over 2 steps) as a white solid.
ESI-MS (m/z): [M+Na]+ 457.91/459.91 (1:1); 1H NMR (400 MHz, CDCl3): δ 7.49 (d, J = 8.4 Hz, 2H), 7.09 (d, J = 8.3 Hz, 2H), 6.55 (s, 1H), 4.41–4.10 (m, 4H), 3.70 (s, 2H), 2.03 (s, 3H), 1.29 (t, J = 7.1 Hz, 6H); 13C NMR (151 MHz, CDCl3): δ 169.4, 167.4, 139.2, 137.2 (t, J = 23.8 Hz), 130.2, 124.5 (t, J = 5.1 Hz), 118.4 (t, J = 303.6 Hz), 67.1, 63.0, 37.7, 23.2, 14.1; 19F NMR (565 MHz, CDCl3) δ –43.15 (s, 2F).
Diethyl 2-acetamido-2-(4-(bromodifluoromethoxy)benzyl)malonate (15)
The title compound was prepared following General Procedure D using crude 11 (44.8 mmol, 1.0 eq), diethylacetamidomalonate (9.72 g, 44.8 mmol, 1.0 eq), K2CO3 (12.4 g, 89.5 mmol, 2.0 eq) and KI (7.43 g, 44.8 mmol, 1.0 eq) in ACN (275 mL). The crude product was purified by silica gel column chromatography (eluent: PE 40/60/EtOAc 75:25) to provide the title compound 15 (8.58 g, 45.1% over 2 steps) as a white solid.
ESI-MS (m/z): [M+Na]+ 473.92/475.91 (1:1); 1H NMR (600 MHz, CDCl3): δ 7.13 (d, J = 8.5 Hz, 2H), 7.04 (d, J = 8.6 Hz, 2H), 6.56 (s, 1H), 4.37–4.17 (m, 4H), 3.66 (s, 2H), 2.03 (s, 3H), 1.28 (t, J = 7.1 Hz, 6H); 13C NMR (151MHz, CDCl3): δ 169.3, 167.5, 150.1 (t, J = 1.7 Hz), 134.4, 131.3, 121.3, 114.6 (t, J = 308.9 Hz), 67.2, 62.9, 37.3, 23.2, 14.1; 19F NMR (565 MHz, CDCl3) δ –16.82 (s, 2F).
Diethyl 2-acetamido-2-(3-(bromodifluoromethoxy)benzyl)malonate (16)
The title compound was prepared following General Procedure D using crude 12 (8.44 mmol, 1.0 eq), diethylacetamidomalonate (1.83 g, 8.44 mmol, 1.0 eq), K2CO3 (2.33 g, 16.9 mmol, 2.0 eq) and KI (1.40 g, 8.44 mmol, 1.0 eq) in ACN (55 mL). The crude product was purified by silica gel column chromatography (eluent: PE 40/60/EtOAc 75:25) to provide the title compound 16 (2.15 g, 56.2% over 2 steps) as a pale-yellow solid.
ESI-MS (m/z): [M+Na]+ 473.96/475.95 (1:1); 1H NMR (400 MHz, CDCl3): δ 7.30 (t, J = 7.9 Hz, 1H), 7.12 (ddd, J = 8.2, 2.2, 0.9 Hz, 1H), 6.97 (d, J = 7.7 Hz, 1H), 6.91 (s, 1H), 4.33–4.18 (m, 4H), 3.68 (s, 3H), 2.03 (s, 3H), 1.29 (t, J = 7.1 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ 169.4, 167.4, 150.9, 137.7, 129.7, 128.7, 122.9, 120.3, 114.6 (t, J = 308.6 Hz), 67.2, 63.0, 37.6, 23.1, 14.1; 19F NMR (376 MHz, CDCl3) δ –15.30 (s, 2F).
Diethyl 2-acetamido-2-(4-((bromodifluoromethyl)thio)benzyl)malonate (17)
The title compound was prepared following General Procedure D using crude 13 (9.63 mmol, 1.0 eq), diethylacetamidomalonate (2.09 g, 9.63 mmol, 1.0 eq), K2CO3 (2.66 g, 19.2 mmol, 2.0 eq) and KI (1.60 g, 9.63 mmol, 1.0 eq) in ACN (55 mL). The crude product was purified by silica gel column chromatography (eluent: PE 40/60/EtOAc 90:10) to provide the title compound 17 (3.47 g, 76.9% over 2 steps) as a pale-yellow solid.
ESI-MS (m/z): [M+Na]+ 489.91/491.89 (1:1); 1H NMR (400 MHz, CDCl3): δ 7.53 (d, J = 8.1 Hz, 2H), 7.07 (d, J = 8.2 Hz, 2H), 6.55 (s, 1H), 4.30–4.21 (m, 4H), 3.68 (s, 2H), 2.02 (s, 3H), 1.27 (t, J = 7.1 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ 169.3, 167.4, 139.0, 136.3, 131.1, 126.1, 119.2 (t, J = 338.4 Hz), 67.1, 63.0, 37.7, 23.1, 14.1; 19F NMR (376 MHz, CDCl3): δ –23.42 (s, 2F).
Diethyl 2-acetamido-2-(4-((bromodifluoromethyl)sulfonyl)benzyl)malonate (18)
To a solution of 17 (200 mg, 0.427 mmol, 1.0 eq) in dry DCM (5mL) was added mCPBA (316 mg, 1.82 mmol, 3.0 eq) at 0 °C. The reaction mixture was stirred at room temperature for 18 h, then additional mCBPA (160 mg, 0.92 mmol, 1.5 eq) and dry DCM (2 mL) were added. After further 22 h of stirring, the solvent was evaporated off in vacuo. The residue was dissolved in EtOAc (50 mL), washed with sat. NaHCO3 (3 × 15 mL), brine (15 mL), dried over Na2SO4 and the solvent was concentrated in vacuo. The crude product was purified by reversed-phase chromatography (C18, eluent: H2O/ACN with 0.1% TFA) and the resulting fractions were freeze-dried to provide the title compound 18 (155.2 mg, 72.6%) as a white solid.
ESI-MS (m/z): [M+H]+ 500.06/502.05 (1:1); 1H NMR (400 MHz, CDCl3): δ 7.92 (d, J = 8.4 Hz, 2H), 7.30 (d, J = 8.5 Hz, 2H), 6.56 (s, 1H), 4.40–4.17 (m, 4H), 3.81 (s, 2H), 2.04 (s, 3H), 1.29 (t, J = 7.1 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ 169.6, 167.1, 145.4, 131.4 (s, 2C), 129.0, 121.1 (t, J = 348.7 Hz), 66.9, 63.3, 38.0, 23.2, 14.2; 19F NMR (376 MHz, CDCl3): δ –57.56 (s, 2F).
2-Acetamido-3-(4-(bromodifluoromethoxy)phenyl)propanoic acid (19)
To a solution of 15 (808 mg, 1.79 mmol, 1.0 eq) in MeOH (12 mL) and H2O (4 mL) was added NaOH (357 g, 8.93 mmol, 40.0 eq) at 0 °C. After 15 min, the reaction mixture stirred at room temperature for 1 h, then at 80 °C for 3 h. MeOH was removed in vacuo, then H2O and Et2O were added and the biphasic mixture was acidified with HCl to pH = 3–5. After that, the mixture was extracted three times with Et2O, the combined organic layers were washed with brine, dried over Na2SO4 and the solvent was removed in vacuo. The crude product was purified by silica gel column chromatography (eluent: DCM/MeOH 98:02 to 86:14 with 0.1% TFA) to provide the title compound 19 (362.8 mg, 57.7%) as a colorless solid.
ESI-MS (m/z): [M-H]− 349.94/351.93 (1:1); 1H NMR (400 MHz, DMSO-d6): δ 7.88 (s, 1H), 7.33 (d, J = 8.3 Hz, 2H), 7.19 (d, J = 8.1 Hz, 2H), 4.33 (s, 1H), 3.12 (dd, J = 13.6, 4.2 Hz, 1H), 2.87 (dd, J = 13.6, 8.5 Hz, 1H), 1.77 (s, 3H); 13C NMR (101 MHz, DMSO-d6): δ 174.6, 169.0, 148.6, 138.3, 130.9, 120.7, 114.4 (t, J = 306.9 Hz), 114.4 (t, J = 306.9 Hz), 54.5, 36.6, 22.6; 19F NMR (376 MHz, DMSO-d6): δ –16.28 (s, 2F).
2-Acetamido-3-(3-(bromodifluoromethoxy)phenyl)propanoic acid (20)
To a solution of 16 (1.82 g, 4.02 mmol, 1.0 eq) in MeOH (45 mL) and H2O (15 mL) was added NaOH (357 g, 8.93 mmol, 40.0 eq) at 0 °C. After 15 min, the reaction mixture stirred at room temperature for 4 h. MeOH was removed in vacuo, then H2O and EtOAc were added and the biphasic mixture was acidified at 0 °C with HCl to pH = 3–5. After that, the mixture was extracted three times with EtOAc and the combined organic layers were washed with brine. The solvent was removed in vacuo and the crude intermediate was dissolved in THF (45 mL) and H2O (15 mL). After 3 h stirring at 70 °C, THF was removed in vacuo and 1 M HCl (40 mL) was added. The aq. phase was extracted three times with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4 and the solvent was concentrated in vacuo to provide the title compound 20 (1.32 g, 93.3%) as a pale-yellow solid.
ESI-MS (m/z): [M-H]− 349.99/351.99 (1:1); 1H NMR (400 MHz, DMSO-d6): δ 8.22 (d, J = 8.2 Hz, 1H), 7.42 (t, J = 8.0 Hz, 1H), 7.28 (d, J = 7.8 Hz, 1H), 7.24–7.13 (m, 2H), 4.43 (ddd, J = 9.8, 8.3, 4.8 Hz, 1H), 3.12 (dd, J = 13.9, 4.8 Hz, 1H), 2.89 (dd, J = 13.9, 9.8 Hz, 1H), 1.77 (s, 3H); 13C NMR (101 MHz, DMSO-d6): δ 172.9, 169.2, 150.0, 140.4, 129.9, 128.2, 121.9, 119.2, 114.3 (t, J = 307.0 Hz, 1H), 53.1, 36.3, 22.3; 19F NMR (376 MHz, DMSO-d6): δ –16.18 (d, 2F).
(S)-2-Amino-3-(4-(bromodifluoromethoxy)phenyl)propanoic acid TFA salt (21)
A solution of 19 (1.59 g, 4.15 mmol) and CoCl2 (1.1 mL, 0.1 M) in 0.1 M K2HPO4/KH2PO4 buffer (220 mL) was adjusted to pH 8. Amano Acylase (56.6 mg, ≥30,000 U g−1, pH 8.0, 50 °C (Optimum pH and temperature)) was added to the solution and the reaction mixture was stirred for 6 h at room temperature, then the mixture was shock-frozen and freeze-dried. The lyophilized residue was suspended in EtOAc and H2O. The biphasic mixture was adjusted to pH 1 by dropwise addition of conc. HCl at 0 °C. The aq. phase was washed three times with EtOAc, the pH was adjusted to pH 5.6 with NaHCO3 and the aq. layer was freeze-dried. The crude product was purified by reversed-phase chromatography (C18, eluent: H2O/ACN with 0.1% TFA) to provide the
HR-ESI-MS (m/z): [M+H]+theor. 309.98849, found 309.98871, rel. Δm 0.72 ppm; 1H NMR (400 MHz, DMSO-d6): δ 7.40 (d, J = 8.6 Hz, 2H), 7.29 (d, J = 8.4 Hz, 2H), 4.09 (t, J = 6.4 Hz, 1H), 3.12 (qd, J = 14.3, 6.5 Hz, 2H); 13C NMR (101 MHz, DMSO-d6): δ 170.4, 149.3, 134.9, 131.4, 121.3, 114.3 (t, J = 307.2 Hz), 53.4, 35.3; 19F NMR (376 MHz, DMSO-d6): δ –18.45 (s, 2F).
(S)-2-Amino-3-(3-(bromodifluoromethoxy)phenyl)propanoic acid TFA salt (22)
A suspension of 20 in ddH2O (220 mL) was adjusted with 0.1 M NaOH to pH 10 until the reactant was completely dissolved. Amano Acylase (220 mg, ≥30,000 U g−1, pH 8.0, 50 °C (Optimum pH and temperature)) was added to the solution and the reaction mixture was stirred for 4 h at room temperature. The pH was kept between 7 and 10 during the reaction. The mixture was freeze-dried and the lyophilized product was purified by reversed-phase chromatography (C18, eluent: H2O/ACN with 0.1% TFA) to provide the
HR-ESI-MS (m/z): [M+H]+theor. 309.98849, found 309.98824, rel. Δm 0.79 ppm; 1H NMR (400 MHz, DMSO-d6): δ 7.46 (t, J = 7.8 Hz, 1H), 7.32 (d, J = 7.7 Hz, 1H), 7.24 (d, J = 9.1 Hz, 2H), 4.01 (t, J = 6.4 Hz, 1H), 3.14 (ddd, J = 21.1, 14.3, 6.4 Hz, 2H); 13C NMR (101 MHz, DMSO-d6): δ 170.0, 150.1, 138.4, 130.2, 128.7, 122.3, 119.7, 117.3, 114.2 (t, J = 307.3 Hz), 53.4, 35.6; 19F NMR (376 MHz, DMSO-d6): δ –18.41 (s, 2F).
(S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(4-(bromodifluoromethoxy)phenyl)propanoic acid (23)
To a solution of 21 (168 mg, 0.396 mmol, 1 eq) in 1,4-dioxane (5 mL) was added at 0 °C a 10% Na2CO3 solution (2 mL, 1.89 mmol, 5 eq), followed by Fmoc-Cl (105 mg, 0.405 mmol, 1.1 eq). After stirring 1 h at 0 °C, the reaction mixture was stirred for 3 h at room temperature, then the mixture was concentrated in vacuo. DCM and H2O were added to the residue and the pH was adjusted at 0 °C with conc. HCl to pH = 1–2. The biphasic mixture was extracted three times with DCM. The combined organic layers were washed with brine, dried over Na2SO4 and the solvent was removed in vacuo. The crude product was purified by reversed-phase chromatography (C18, eluent: H2O and ACN with 0.1% TFA) to provide the title compound 23 (98.0 mg, 46.5%) as a white solid.
HR-ESI-MS (m/z): [M-H]−theor. 530.04201, found 530.04280, rel. Δm 1.48 ppm; 1H NMR (700 MHz, DMSO-d6): δ 12.80 (bs, 1H), 7.88 (d, J = 7.5 Hz, 2H), 7.78 (d, J = 8.6 Hz, 1H), 7.64 (t, J = 6.9 Hz, 2H), 7.43–7.38 (m, 4H), 7.33–7.27 (m, 2H), 7.22 (d, J = 8.4 Hz, 2H), 4.24–4.12 (m, 4H), 3.12 (dd, J = 13.9, 4.3 Hz, 1H), 2.90 (dd, J = 13.8, 10.8 Hz, 1H); 13C NMR (176 MHz, DMSO-d6): δ 173.6, 156.4, 149.2, 144.2 (d, J = 6.5 Hz), 141.2 (d, J = 5.0 Hz), 138.0, 131.3, 128.1, 127.5, 125.7 (d, J = 7.4 Hz), 121.4, 120.6, 114.8 (t, J = 306.9 Hz), 66.1, 55.7, 47.0, 36.2; 19F NMR (659 MHz, DMSO-d6): δ –15.92 (d, J = 7.7 Hz, 2F).
(S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(3-(bromodifluoromethoxy)phenyl)propanoic acid (24)
To a solution of 22 (156 mg, 0.368 mmol, 1 eq) in 1,4-dioxane (5 mL) was added at 0 °C a 10% Na2CO3 solution (2 mL, 1.89 mmol, 5 eq), followed by Fmoc-Cl (105 mg, 0.405 mmol, 1.1 eq). The reaction mixture was stirred for 4 h at room temperature. H2O was added and the mixture was washed with Et2O, then EtOAc was added to the aq. layer and the pH was adjusted at 0 °C with conc. HCl to pH = 1–2. The biphasic mixture was extracted three times with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4 and the solvent was removed in vacuo. The crude product was purified by reversed-phase chromatography (C18, eluent: H2O and ACN with 0.1% TFA) to provide the title compound 24 (176.2 mg, 89.9%) as a white solid.
HR-ESI-MS (m/z): [M+Na]+theor. 554.03851, found 554.03874, rel. Δm 0.41 ppm; 1H NMR (700 MHz, DMSO-d6): δ 12.82 (bs, 1H), 7.88 (d, J = 7.6 Hz, 2H), 7.78 (d, J = 8.6 Hz, 1H), 7.63 (dd, J = 8.9, 8.3 Hz, 2H), 7.45–7.36 (m, 3H), 7.35–7.23 (m, 4H), 7.19 (d, J = 8.1 Hz, 1H), 4.24–4.13 (m, 4H), 3.16 (dd, J = 13.8, 4.2 Hz, 1H), 2.93 (dd, J = 13.8, 10.9 Hz, 1H); 13C NMR (176 MHz, DMSO-d6): δ 173.5, 156.4, 150.5, 144.2, 141.2, 141.1, 130.4, 128.8, 128.1, 127.5, 125.7 (d, J = 11.9 Hz), 122.5, 120.6, 119.7, 114.8 (t, J = 307.1 Hz), 66.1, 55.6, 47.0, 36.4; 19F NMR (659 MHz, DMSO-d6): δ –15.76 (s, 2F).
N2-(((9H-Fluoren-9-yl)methoxy)carbonyl)-N6-(2,2-difluoroacetyl)-
The title compound was prepared following General Procedure E using Fmoc-Lys-OH (3.0 g, 8.14 mmol, 1 eq), NMM (2.69 mL, 24.4 mmol, 0.92 g cm−3, 3 eq) and 2,2-difluoroacetic anhydride (1.70 g, 9.77 mmol, 1.2 eq) in dry THF (60 mL). The crude product was purified by reversed-phase chromatography (eluent: H2O/ACN with 0.1% TFA) to provide the title compound 25b (3.27 g, 89.8%) as a white solid.
ESI-MS (m/z): [M-H]− 445.19; 1H NMR (400 MHz, DMSO-d6): δ 8.78 (t, J = 5.3 Hz, 1H), 7.89 (d, J = 7.5 Hz, 2H), 7.73 (d, J = 7.4 Hz, 2H), 7.63 (d, J = 8.0 Hz, 1H), 7.42 (t, J = 7.3 Hz, 2H), 7.33 (td, J = 7.4, 0.8 Hz, 2H), 6.18 (t, J = 53.8 Hz, 1H), 4.29 (d, J = 6.6 Hz, 2H), 4.26–4.17 (m, 1H), 3.92 (td, J = 9.3, 4.7 Hz, 1H), 3.13 (dd, J = 13.0, 6.5 Hz, 2H), 1.81–1.54 (m, 2H), 1.53–1.20 (m, 4H); 13C NMR (101 MHz, DMSO-d6): δ 174.0, 162.1 (t, J = 25.0 Hz), 156.2, 143.9 (d, J = 4.2 Hz), 140.8, 127.7, 127.1, 125.3, 120.1, 108.6 (t, J = 246.7 Hz, 65.6, 53.8, 46.7, 38.3, 30.4, 28.2, 23.0; 19F NMR (376 MHz, DMSO-d6): δ –125.68 (d, J = 53.8 Hz, 2F).
N2-(((9H-Fluoren-9-yl)methoxy)carbonyl)-N6-(2-chloro-2,2-difluoroacetyl)-
The title compound was prepared following General Procedure E using Fmoc-Lys-OH (3.0 g, 8.14 mmol, 1 eq), NMM (2.69 mL, 24.4 mmol, 0.92 g cm−3, 3 eq) and 2-chloro-2,2-difluoroacetic anhydride (2.37 g, 9.77 mmol, 1.2 eq) in dry THF (60 mL). The crude product was purified by reversed-phase chromatography (eluent: H2O/ACN with 0.1% TFA) to provide the title compound 25d (2.18 g, 55.7%) as a white solid.
ESI-MS (m/z): [M+Na]+ 503.17; 1H NMR (400 MHz, DMSO-d6): δ 9.25 (t, J = 5.4 Hz, 1H), 7.89 (d, J = 7.5 Hz, 2H), 7.73 (d, J = 7.4 Hz, 2H), 7.63 (d, J = 8.0 Hz, 1H), 7.42 (t, J = 7.3 Hz, 2H), 7.33 (td, J = 7.4, 1.0 Hz, 2H), 4.39–4.27 (m, 2H), 4.27–4.15 (m, 1H), 3.92 (td, J = 9.4, 4.7 Hz, 1H), 3.17 (dd, J = 13.3, 6.3 Hz, 2H), 1.81–1.25 (m, 6H); 13C NMR (101 MHz, DMSO-d6): δ 174.0, 158.6 (t, J = 29.4 Hz), 156.2, 143.8 (d, J = 4.9 Hz), 140.7, 127.7, 127.1, 125.3, 120.1, 119.1 (t, J = 302.6 Hz), 65.6, 53.7, 46.7, 39.2, 30.3, 27.9, 22.9; 19F NMR (376 MHz, DMSO-d6): δ –62.73 (s, 2F).
N2-(((9H-Fluoren-9-yl)methoxy)carbonyl)-N6-(2-bromo-2,2-difluoroacetyl)-
The title compound was prepared following General Procedure E using Fmoc-Lys-OH (3.0 g, 8.14 mmol, 1 eq), NMM (2.69 mL, 24.4 mmol, 0.92 g cm−3, 3 eq) and 2-bromo-2,2-difluoroacetic anhydride (3.24 g, 9.77 mmol, 1.2 eq) in dry THF (60 mL). The crude product was purified by reversed-phase chromatography (eluent: H2O/ACN with 0.1% TFA) to provide the title compound 25e (2.44 g, 57.0%) as a white solid.
ESI-MS (m/z): [M+H]+ 523.10/525.10 (1:1); 1H NMR (400 MHz, DMSO-d6): δ 9.18 (t, J = 5.5 Hz, 1H), 7.89 (d, J = 7.5 Hz, 2H), 7.73 (d, J = 7.4 Hz, 2H), 7.63 (d, J = 8.0 Hz, 1H), 7.42 (t, J = 7.5 Hz, 2H), 7.33 (td, J = 7.4, 1.0 Hz, 2H), 4.28 (dd, J = 9.9, 3.3 Hz, 2H), 4.25–4.18 (m, 1H), 3.92 (td, J = 9.4, 4.7 Hz, 1H), 3.16 (dd, J = 13.5, 6.3 Hz, 2H), 1.78–1.29 (m, 6H); 13C NMR (101 MHz, DMSO-d6): δ 173.9, 159.5 (t, J = 26.9 Hz), 156.2, 143.8 (d, J = 4.9 Hz), 143.8 (d, J = 4.9 Hz), 140.7, 127.6, 127.1, 125.3, 120.1, 111.9 (t, J = 315.1 Hz), 65.6, 53.7, 46.7, 39.1, 30.3, 27.9, 22.9; 19F NMR (376 MHz, DMSO-d6): δ –59.92 (s, 2F).
(S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-acetamidobutanoic acid (26a)
The title compound was prepared following General Procedure E using Fmoc-Dab-OH (1.0 g, 2.94 mmol, 1 eq), NMM (970 µL, 8.82 mmol, 0.92 g cm−3, 3 eq) and acetic anhydride (360 mg, 3.53 mmol, 1.2 eq) in dry THF (20 mL). The crude product was purified by reversed-phase chromatography (eluent: H2O/ACN with 0.1% TFA) to provide the title compound 26a (498.9 mg, 45.0%) as a white solid.
ESI-MS (m/z): [M+H]− 381.06; 1H NMR (400 MHz, DMSO-d6): δ 7.90 (d, J = 7.5 Hz, 2H), 7.78 (d, J = 8.5 Hz, 1H), 7.71 (d, J = 7.4 Hz, 2H), 7.42 (t, J = 7.4 Hz, 2H), 7.34 (t, J = 7.1 Hz, 2H), 4.48–4.29 (m, 3H), 4.25 (t, J = 6.7 Hz, 1H), 3.73 (t, J = 10.0 Hz, 1H), 3.40 (td, J = 10.9, 7.0 Hz, 1H), 2.37 (s, 1H), 2.28–2.13 (m, 1H), 1.88 (dt, J = 22.5, 11.2 Hz, 1H); 13C NMR (101 MHz, DMSO-d6): δ 173.5, 170.2, 155.8, 143.8, 140.8, 127.6, 127.1, 125.1, 120.1, 65.7, 53.2, 46.6, 40.9, 24.4, 23.7.
(S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-(2,2-difluoroacetamido)butanoic acid (26b)
The title compound was prepared following General Procedure E using Fmoc-Dab-OH (1.0 g, 2.94 mmol, 1 eq), NMM (970 µL, 8.82 mmol, 0.92 g cm−3, 3 eq) and 2,2-difluoroacetic anhydride (609 mg, 3.53 mmol, 1.2 eq) in dry THF (20 mL). The crude product was purified by reversed-phase chromatography (eluent: H2O/ACN with 0.1% TFA) to provide the title compound 26b (298.6 mg, 24.6%) as a white solid.
ESI-MS (m/z): [M+Na]+ 441.07; 1H NMR (400 MHz, DMSO-d6): δ 12.63 (bs, 1H), 8.83 (t, J = 5.2 Hz, 1H), 7.89 (d, J = 7.5 Hz, 2H), 7.73 (dd, J = 7.7, 4.6 Hz, 3H), 7.41 (dt, J = 7.5, 3.7 Hz, 2H), 7.38–7.28 (m, 2H), 6.19 (t, J = 53.7 Hz, 1H), 4.33–4.20 (m, 3H), 4.01 (td, J = 9.0, 4.7 Hz, 1H), 3.23 (dd, J = 12.9, 6.6 Hz, 2H), 1.98 (td, J = 12.8, 7.6 Hz, 1H), 1.86–1.73 (m, 1H); 13C NMR (101 MHz, DMSO-d6): δ 173.6, 162.3 (t, J = 25.1 Hz), 156.2, 143.8, 140.7, 127.7, 127.1, 125.3 (d, J = 3.3 Hz), 120.1, 108.5 (t, J = 246.6 Hz, 65.7, 51.6, 46.7, 35.8, 30.0; 19F NMR (376 MHz, DMSO-d6): δ –125.76 (d, J = 53.7 Hz, 2F).
(S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-(2,2,2-trifluoroacetamido)butanoic acid (26c)
The title compound was prepared following General Procedure E using Fmoc-Dab-OH (1.0 g, 2.94 mmol, 1 eq), NMM (970 µL, 8.82 mmol, 0.92 g cm−3, 3 eq) and 2,2,2-trifluoroacetic anhydride (741 mg, 3.53 mmol, 1.2 eq) in dry THF (20 mL). The crude product was purified by reversed-phase chromatography (eluent: H2O/ACN with 0.1 TFA) to provide the title compound 26c (273.0 mg, 21.6%) as a white solid.
ESI-MS (m/z): [M+Na]+ 459.05; 1H NMR (400 MHz, DMSO-d6): δ 12.73 (bs, 1H), 9.47 (t, J = 5.2 Hz, 1H), 7.91 (d, J = 7.5 Hz, 2H), 7.75 (d, J = 6.7 Hz, 3H), 7.44 (t, J = 7.3 Hz, 2H), 7.35 (t, J = 7.4 Hz, 2H), 4.36–4.19 (m, 3H), 4.03 (td, J = 9.4, 4.7 Hz, 1H), 3.30 (m, 2H), 2.10–1.97 (m, 1H), 1.92–1.77 (m, 1H); 13C NMR (101 MHz, DMSO-d6): δ 173.4, 156.8–155.6 (m, 2C) 143.8, 140.7, 127.6, 127.1, 125.3 (d, J = 4.4 Hz), 120.2–111.6 (m, 2C), 65.7, 51.5, 46.6, 36.4, 29.6; 19F NMR (376 MHz, DMSO-d6): δ –74.76 (s, 3F).
(S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-(2-chloro-2,2-difluoroacetamido)butanoic acid (26d)
The title compound was prepared following General Procedure E using Fmoc-Dab-OH (1.0 g, 2.94 mmol, 1 eq), NMM (970 µL, 8.82 mmol, 0.92 g cm−3, 3 eq) and 2-chloro-2,2-difluoroacetic anhydride (857 mg, 3.53 mmol, 1.2 eq) in dry THF (20 mL). The crude product was purified by reversed-phase chromatography (eluent: H2O/ACN with 0.1% TFA) to provide the title compound 26d (306.2 mg, 23.3%) as a white solid.
ESI-MS (m/z): [M+Na]+ 475.03/477.02 (3:1); 1H NMR (400 MHz, DMSO-d6): δ 12.65 (bs, 1H), 9.29 (t, J = 5.3 Hz, 1H), 7.89 (d, J = 7.5 Hz, 2H), 7.72 (dd, J = 7.8, 3.7 Hz, 3H), 7.42 (t, J = 7.3 Hz, 2H), 7.38–7.28 (m, 2H), 4.33–4.18 (m, 3H), 4.01 (td, J = 9.4, 4.7 Hz, 1H), 3.27 (dd, J = 13.3, 7.3 Hz, 2H), 2.01 (qd, J = 7.7, 4.9 Hz, 1H), 1.88–1.74 (m, 1H); 13C NMR (101 MHz, DMSO-d6): δ 173.4, 158.7 (t, J = 29.5 Hz), 156.1, 143.8, 140.7, 127.7, 127.1, 125.3 (d, J = 4.3 Hz), 120.1, 119.0 (t, J = 302.6 Hz), 65.7, 51.5, 46.6, 36.7, 29.7; 19F NMR (376 MHz, DMSO-d6): δ –62.72 (s, 2F).
(S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-(2-bromo-2,2-difluoroacetamido)butanoic acid (26e)
The title compound was prepared following General Procedure E using Fmoc-Dab-OH (1.0 g, 2.94 mmol, 1 eq), NMM (970 µL, 8.82 mmol, 0.92 g cm−3, 3 eq) and 2-bromo-2,2-difluoroacetic anhydride (1.17 g, 3.53 mmol, 1.2 eq) in dry THF (20 mL). The crude product was purified by reversed-phase chromatography (eluent: H2O/ACN with 0.1% TFA) to provide the title compound 26e (341.7 mg, 23.7%) as a white solid.
ESI-MS (m/z): [M+Na]+ 518.97/520.97 (1:1); 1H NMR (400 MHz, DMSO-d6): δ 12.70 (s, 1H), 9.23 (t, J = 5.2 Hz, 1H), 7.89 (d, J = 7.5 Hz, 2H), 7.73 (dd, J = 7.7, 3.9 Hz, 3H), 7.42 (t, J = 7.2 Hz, 2H), 7.33 (ddd, J = 7.4, 4.4, 1.3 Hz, 2H), 4.37–4.15 (m, 4H), 4.01 (td, J = 8.9, 4.7 Hz, 1H), 3.26 (dd, J = 12.8, 6.4 Hz, 2H), 2.01 (td, J = 12.8, 7.5 Hz, 1H), 1.81 (tt, J = 10.1, 5.0 Hz, 1H); 13C NMR (101 MHz, DMSO-d6): δ 173.5, 159.6 (t, J = 27.0 Hz), 156.1, 143.8, 140.7, 127.7, 127.1, 125.3 (d, J = 4.4 Hz), 120.1, 111.8 (t, J = 314.9 Hz), 65.7, 51.5, 46.6, 36.6, 29.7; 19F NMR (376 MHz, DMSO-d6): δ –59.97 (s, 2F).
Abbreviations
Abu, 2-aminobutanoic acid; β-ME, 2-mercaptoethanol; Dab, 2.4-diaminobutyric acid; FP, fluorescence polarization; HB, hydrogen bond; Mdm2, mouse double minute 2 homolog; Mdm4, mouse double minute 4 homolog; Nle, norleucine; PDB, protein data bank; PPI, protein–protein interaction; SD, standard deviation; SPPS, solid-phase peptide synthesis; XB, halogen bond.
Acknowledgments
We thank Prof. Dr. Harald Groß for the permission to record CD spectra and Jun.-Prof. Matthias Gehringer for useful discussions. The authors acknowledge support from the state of Baden–Württemberg through bwHPC and the German Research Foundation (DFG) through Grant No. INST 40/575-1 FUGG (JUSTUS 2 cluster). In addition, support is acknowledged from the High Performance and Cloud Computing Group at the Zentrum für Datenverarbeitung of the University of Tübingen and the German Research Foundation (DFG) through Grant No. INST 37/935-1 FUGG (BinAC cluster).
Author Contributions
All authors made a significant contribution to the work reported: F.M.B. envisioned the research. S.V. and F.M.B. conceptualized the experiments and designed the study. S.V. performed the synthesis, analytical and biophysical work. T.K. prepared the protein by heterologous expression. M.O.Z. performed all computational investigations, as well as data analysis of the results. S.V. conducted data analysis and reprocessing of the experimental data. S.V., M.O.Z. and F.M.B. wrote the manuscript. T.K. critically reviewed the manuscript. All authors have agreed on the journal to which the article has been submitted. All authors agreed on all versions of the article at any stage of the submission, revision, publication and proofing process, particularly including the final version accepted for publication. All authors agree to take responsibility and be accountable for the contents of the article.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Mardirossian M, Rubini M, Adamo MFA, et al. Natural and synthetic halogenated amino acids-structural and bioactive features in antimicrobial peptides and peptidomimetics. Molecules. 2021;26(23):7401. doi:10.3390/molecules26237401
2. Scholfield MR, Ford MC, Carlsson A-C-C, Butta H, Mehl RA, Ho PS. Structure-energy relationships of halogen bonds in proteins. Biochemistry. 2017;56(22):2794–2802. doi:10.1021/acs.biochem.7b00022
3. Carlsson A-C-C, Scholfield MR, Rowe RK, et al. Increasing enzyme stability and activity through hydrogen bond-enhanced halogen bonds. Biochemistry. 2018;57(28):4135–4147. doi:10.1021/acs.biochem.8b00603
4. Erdélyi M. Halogen bonding in solution. Chem Soc Rev. 2012;41(9):3547–3557. doi:10.1039/C2CS15292D
5. Danelius E, Andersson H, Jarvoll P, Lood K, Gräfenstein J, Erdélyi M. Halogen bonding: a powerful tool for modulation of peptide conformation. Biochemistry. 2017;56(25):3265–3272. doi:10.1021/acs.biochem.7b00429
6. Erdélyi M. Application of the halogen bond in protein systems. Biochemistry. 2017;56(22):2759–2761. doi:10.1021/acs.biochem.7b00371
7. Peintner S, Erdélyi M. Pushing the limits of characterising a weak halogen bond in solution. Chem Eur J. 2022;28(5):e202103559. doi:10.1002/chem.202103559
8. Bertolani A, Pirrie L, Stefan L, et al. Supramolecular amplification of amyloid self-assembly by iodination. Nat Commun. 2015;6:7574. doi:10.1038/ncomms8574
9. Pizzi A, Dichiarante V, Terraneo G, Metrangolo P. Crystallographic insights into the self-assembly of KLVFF amyloid-beta peptides. Pept Sci. 2018;100:e23088. doi:10.1002/bip.23088
10. Pizzi A, Lascialfari L, Demitri N, et al. Halogen bonding modulates hydrogel formation from fmoc amino acids. CrystEngComm. 2017;19(14):1870–1874. doi:10.1039/C7CE00031F
11. Bergamaschi G, Lascialfari L, Pizzi A, et al. A halogen bond-donor amino acid for organocatalysis in water. Chem Comm. 2018;54(76):10718–10721. doi:10.1039/C8CC06010J
12. Pizzi A, Demitri N, Terraneo G, Metrangolo P. Halogen bonding at the wet interfaces of an amyloid peptide structure. CrystEngComm. 2018;20(36):5321–5326. doi:10.1039/C8CE01205A
13. Pizzi A, Catalano L, Demitri N, Dichiarante V, Terraneo G, Metrangolo P. Halogen bonding as a key interaction in the self‐assembly of iodinated diphenylalanine peptides. Pept Sci. 2020;112(1):e24127. doi:10.1002/pep2.24127
14. Pizzi A, Pigliacelli C, Bergamaschi G, Gori A, Metrangolo P. Biomimetic engineering of the molecular recognition and self-assembly of peptides and proteins via halogenation. Coord Chem Rev. 2020;411:213242. doi:10.1016/j.ccr.2020.213242
15. Marchetti A, Pizzi A, Bergamaschi G, et al. Fibril structure demonstrates the role of iodine labelling on a pentapeptide self-assembly. Chem Eur J. 2022;28(14):e202104089. doi:10.1002/chem.202104089
16. Bittner S, Scherzer R, Harlev E. The five bromotryptophans. Amino Acids. 2007;33(1):19–42. doi:10.1007/s00726-006-0441-8
17. Strickland M, Willis CL. Synthesis of halogenated α‐amino acids. In: Hughes AB, editor. Amino Acids, Peptides and Proteins in Organic Chemistry: Origins and Synthesis of Amino Acids. Vol. 1. Weinheim: Wiley-VCH; 2009:441–471.
18. Erak M, Bellmann-Sickert K, Els-Heindl S, Beck-Sickinger AG. Peptide chemistry toolbox - transforming natural peptides into peptide therapeutics. Bioorg Med Chem. 2018;26(10):2759–2765. doi:10.1016/j.bmc.2018.01.012
19. Li W, Separovic F, O’Brien-Simpson NM, Wade JD. Chemically modified and conjugated antimicrobial peptides against superbugs. Chem Soc Rev. 2021;50(8):4932–4973. doi:10.1039/d0cs01026j
20. Xie M, Liu D, Yang Y. Anti-cancer peptides: classification, mechanism of action, reconstruction and modification. Open Biol. 2020;10(7):200004. doi:10.1098/rsob.200004
21. Hardegger LA, Kuhn B, Spinnler B, et al. Systematic investigation of halogen bonding in protein-ligand interactions. Angew Chem Int Ed. 2011;50(1):314–318. doi:10.1002/anie.201006781
22. Parisini E, Metrangolo P, Pilati T, Resnati G, Terraneo G. Halogen bonding in halocarbon-protein complexes: a structural survey. Chem Soc Rev. 2011;40(5):2267–2278. doi:10.1039/c0cs00177e
23. Clark T, Hennemann M, Murray JS, Politzer P. Halogen bonding: the sigma-hole. Proceedings of “modeling interactions in biomolecules II”, Prague, September 5th-9th, 2005. J Mol Model. 2007;13(2):291–296. doi:10.1007/s00894-006-0130-2
24. Wilcken R, Liu X, Zimmermann MO, et al. Halogen-enriched fragment libraries as leads for drug rescue of mutant p53. J Am Chem Soc. 2012;134(15):6810–6818. doi:10.1021/ja301056a
25. Wilcken R, Zimmermann MO, Lange A, Zahn S, Boeckler FM. Using halogen bonds to address the protein backbone: a systematic evaluation. J Comput Aided Mol Des. 2012;26(8):935–945. doi:10.1007/s10822-012-9592-8
26. Lange A, Heidrich J, Zimmermann MO, Exner TE, Boeckler FM. Scaffold effects on halogen bonding strength. J Chem Inf Model. 2019;59(2):885–894. doi:10.1021/acs.jcim.8b00621
27. Zimmermann MO, Boeckler FM. Targeting the protein backbone with aryl halides: systematic comparison of halogen bonding and π⋯π interactions using N-methylacetamide. Medchemcomm. 2016;7(3):500–505. doi:10.1039/C5MD00499C
28. Zimmermann MO, Lange A, Boeckler FM. Evaluating the potential of halogen bonding in molecular design: automated scaffold decoration using the new scoring function XBScore. J Chem Inf Model. 2015;55(3):687–699. doi:10.1021/ci5007118
29. Zimmermann MO, Lange A, Zahn S, Exner TE, Boeckler FM. Using surface scans for the evaluation of halogen bonds toward the side chains of aspartate, asparagine, glutamate, and glutamine. J Chem Inf Model. 2016;56(7):1373–1383. doi:10.1021/acs.jcim.6b00075
30. Lange A, Zimmermann MO, Wilcken R, Zahn S, Boeckler FM. Targeting histidine side chains in molecular design through nitrogen-halogen bonds. J Chem Inf Model. 2013;53(12):3178–3189. doi:10.1021/ci4004305
31. Wilcken R, Zimmermann MO, Lange A, Zahn S, Kirchner B, Boeckler FM. Addressing methionine in molecular design through directed sulfur-halogen bonds. J Chem Theory Comput. 2011;7(7):2307–2315. doi:10.1021/ct200245e
32. Katagiri T, Handa M, Matsukawa Y, Dileep Kumar JS, Uneyama K. Efficient synthesis of an optically pure β-bromo-β,β-difluoroalanine derivative, a general precursor for β,β-difluoroamino acids. Tetrahedron Asymmetry. 2001;12(9):1303–1311. doi:10.1016/S0957-4166(01)00237-3
33. Suzuki A, Mae M, Amii H, Uneyama K. Catalytic route to the synthesis of optically active beta,beta-difluoroglutamic acid and beta,beta-difluoroproline derivatives. J Org Chem. 2004;69(15):5132–5134. doi:10.1021/jo049789c
34. Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 2008;77(1):557–582. doi:10.1146/annurev.biochem.77.060806.091238
35. Kirsch DG, Kastan MB. Tumor-suppressor p53: implications for tumor development and prognosis. J Clin Oncol. 1998;16(9):3158–3168. doi:10.1200/JCO.1998.16.9.3158
36. Boeckler FM, Joerger AC, Jaggi G, Rutherford TJ, Veprintsev DB, Fersht AR. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc Natl Acad Sci. 2008;105(30):10360–10365. doi:10.1073/pnas.0805326105
37. Wilcken R, Wang G, Boeckler FM, Fersht AR. Kinetic mechanism of p53 oncogenic mutant aggregation and its inhibition. Proc Natl Acad Sci. 2012;109(34):13584–13589. doi:10.1073/pnas.1211550109
38. Vogel SM, Bauer MR, Joerger AC, et al. Lithocholic acid is an endogenous inhibitor of MDM4 and MDM2. Proc Natl Acad Sci. 2012;109(42):16906–16910. doi:10.1073/pnas.1215060109
39. Abdel-Halim M, Keeton AB, Gurpinar E, et al. Trisubstituted and tetrasubstituted pyrazolines as a novel class of cell-growth inhibitors in tumor cells with wild type p53. Bioorg Med Chem. 2013;21(23):7343–7356. doi:10.1016/j.bmc.2013.09.055
40. Wilcken R, Zimmermann MO, Bauer MR, et al. Experimental and theoretical evaluation of the ethynyl moiety as a halogen bioisostere. ACS Chem Biol. 2015;10(12):2725–2732. doi:10.1021/acschembio.5b00515
41. Joerger AC, Bauer MR, Wilcken R, et al. Exploiting transient protein states for the design of small-molecule stabilizers of mutant p53. Structure. 2015;23(12):2246–2255. doi:10.1016/j.str.2015.10.016
42. Bauer MR, Jones RN, Baud MGJ, et al. Harnessing fluorine-sulfur contacts and multipolar interactions for the design of p53 mutant Y220C rescue drugs. ACS Chem Biol. 2016;11(8):2265–2274. doi:10.1021/acschembio.6b00315
43. Stahlecker J, Klett T, Schwer M, et al. Revisiting a challenging p53 binding site: a diversity-optimized HEFLib reveals diverse binding modes in T-p53C-Y220C. RSC Med Chem. 2022;13(12):1575–1586. doi:10.1039/D2MD00246A
44. Li Z, Zhao H, Wan C. Cyclized Helical Peptides. Weinheim: Wiley-VCH; 2021.
45. Bell S, Klein C, Müller L, Hansen S, Buchner J. P53 contains large unstructured regions in its native state. J Mol Biol. 2002;322(5):917–927. doi:10.1016/S0022-2836(02)00848-3
46. Dawson R, Müller L, Dehner A, Klein C, Kessler H, Buchner J. The N-terminal domain of p53 is natively unfolded. J Mol Biol. 2003;332(5):1131–1141. doi:10.1016/j.jmb.2003.08.008
47. Kussie PH, Gorina S, Marechal V, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274(5289):948–953. doi:10.1126/science.274.5289.948
48. Popowicz GM, Czarna A, Holak TA. Structure of the human Mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle. 2008;7(15):2441–2443. doi:10.4161/cc.6365
49. Pazgier M, Liu M, Zou G, et al. Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. Proc Natl Acad Sci. 2009;106(12):4665–4670. doi:10.1073/pnas.0900947106
50. Li C, Pazgier M, Li C, et al. Systematic mutational analysis of peptide inhibition of the p53-MDM2/MDMX interactions. J Mol Biol. 2010;398(2):200–213. doi:10.1016/j.jmb.2010.03.005
51. Nikolovska-Coleska Z, Wang R, Fang X, et al. Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal Biochem. 2004;332(2):261–273. doi:10.1016/j.ab.2004.05.055
52. Czarna A, Popowicz GM, Pecak A, Wolf S, Dubin G, Holak TA. High affinity interaction of the p53 peptide-analogue with human Mdm2 and Mdmx. Cell Cycle. 2009;8(8):1176–1184. doi:10.4161/cc.8.8.8185
53. Schon O, Friedler A, Bycroft M, Freund SMV, Fersht AR. Molecular mechanism of the interaction between MDM2 and p53. J Mol Biol. 2002;323(3):491–501. doi:10.1016/S0022-2836(02)00852-5
54. Sánchez-Puig N, Veprintsev DB, Fersht AR. Binding of natively unfolded HIF-1alpha ODD domain to p53. Mol Cell. 2005;17(1):11–21. doi:10.1016/j.molcel.2004.11.019
55. Grimme S, Antony J, Ehrlich S, Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys. 2010;132(15):154104. doi:10.1063/1.3382344
56. Schäfer A, Horn H, Ahlrichs R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J Chem Phys. 1992;97(4):2571–2577. doi:10.1063/1.463096
57. Tao J, Perdew JP, Staroverov VN, Scuseria GE. Climbing the density functional ladder: nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys Rev Lett. 2003;91(14):146401. doi:10.1103/PhysRevLett.91.146401
58. Weigend F, Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys Chem Chem Phys. 2005;7(18):3297–3305. doi:10.1039/B508541A
59. TURBOMOLE V7.4.1 2019, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH; 2007. Available from http://www.turbomole.com.
60. Weber IT, Wu J, Adomat J, et al. Crystallographic analysis of human immunodeficiency virus 1 protease with an analog of the conserved CA-p2 substrate – interactions with frequently occurring glutamic acid residue at P2’ position of substrates. Eur J Biochem. 1997;249(2):523–530. doi:10.1111/j.1432-1033.1997.00523.x
61. Jung JH, Bae S, Lee JY, et al. E3 ubiquitin ligase Hades negatively regulates the exonuclear function of p53. Cell Death Differ. 2011;18(12):1865–1875. doi:10.1038/cdd.2011.57
62. Zhan YA, Wu H, Powell AT, Daughdrill GW, Ytreberg FM. Impact of the K24N mutation on the transactivation domain of p53 and its binding to murine double-minute clone 2. Proteins. 2013;81(10):1738–1747. doi:10.1002/prot.24310
63. Mizuta S, Stenhagen ISR, O’Duill M, et al. Catalytic decarboxylative fluorination for the synthesis of tri- and difluoromethyl arenes. Org Lett. 2013;15(11):2648–2651. doi:10.1021/ol4009377
64. Verhoog S, Pfeifer L, Khotavivattana T, et al. Silver-mediated 18F-labeling of aryl-CF3 and aryl-CHF2 with 18F-fluoride. Synlett. 2015;27(01):25–28. doi:10.1055/s-0035-1560592
65. Barton DHR, Crich D, Motherwell WB. The invention of new radical chain reactions. Part VIII. Radical chemistry of thiohydroxamic esters; a new method for the generation of carbon radicals from carboxylic acids. Tetrahedron. 1985;41(19):3901–3924. doi:10.1016/S0040-4020(01)97173-X
66. Khotavivattana T, Verhoog S, Tredwell M, et al. 18F-labeling of aryl-SCF3, -OCF3 and -OCHF2 with [18F]fluoride. Angew Chem Int Ed. 2015;54(34):9991–9995. doi:10.1002/anie.201504665
67. Burton DJ, Denise MW. Synthesis of bromodifluoromethyl phenyl sulfide, sulfoxide and sulfone. J Fluor Chem. 1981;18(4):573–582. doi:10.1016/S0022-1139(00)82673-1
68. Wang Y-S, Fang X, Chen H-Y, et al. Genetic incorporation of twelve meta-substituted phenylalanine derivatives using a single pyrrolysyl-tRNA synthetase mutant. ACS Chem Biol. 2013;8(2):405–415. doi:10.1021/cb300512r
69. Pearson DA, Blanchette M, Baker ML, Guindon CA. Trialkylsilanes as scavengers for the trifluoroacetic acid deblocking of protecting groups in peptide synthesis. Tetrahedron Lett. 1989;30(21):2739–2742. doi:10.1016/S0040-4039(00)99113-5
70. Popowicz GM, Czarna A, Rothweiler U, et al. Molecular basis for the inhibition of p53 by Mdmx. Cell Cycle. 2007;6(19):2386–2392. doi:10.4161/cc.6.19.4740
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.