")
Back to Journals » Journal of Inflammation Research » Volume 17
The Role of Immune Cells in DKD: Mechanisms and Targeted Therapies
Authors Peng QY, An Y, Jiang ZZ, Xu Y
Received 2 January 2024
Accepted for publication 19 March 2024
Published 6 April 2024 Volume 2024:17 Pages 2103—2118
DOI https://doi.org/10.2147/JIR.S457526
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Qiu-Yue Peng,1– 3,* Ying An,1– 3,* Zong-Zhe Jiang,1– 3 Yong Xu1– 3
1Department of Endocrinology and Metabolism, the Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan, People’s Republic of China; 2Metabolic Vascular Disease Key Laboratory of Sichuan Province, Sichuan, People’s Republic of China; 3Sichuan Clinical Research Center for Nephropathy, Luzhou, Sichuan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yong Xu; Zong-zhe Jiang, Email [email protected]; [email protected]
Abstract: Diabetic kidney disease (DKD), is a common microvascular complication and a major cause of death in patients with diabetes. Disorders of immune cells and immune cytokines can accelerate DKD development of in a number of ways. As the kidney is composed of complex and highly differentiated cells, the interactions among different cell types and immune cells play important regulatory roles in disease development. Here, we summarize the latest research into the molecular mechanisms underlying the interactions among various immune and renal cells in DKD. In addition, we discuss the most recent studies related to single cell technology and bioinformatics analysis in the field of DKD. The aims of our review were to explore immune cells as potential therapeutic targets in DKD and provide some guidance for future clinical treatments.
Keywords: diabetic kidney disease, immune cells, bioinformatics analysis, single-cell analysis
Introduction
The prevalence of diabetes in people aged between 20 and 79 years worldwide was forecast to be 10.5% in 2021 and is expected to rise to 12.2% by 2045.1–4 Diabetic kidney disease (DKD), a chronic kidney disease resulting from diabetes, is defined by persistent proteinuria and ongoing deterioration in renal function, and affects 20% to 50% of patients with diabetes.5 DKD is the main cause of end-stage renal disease (ESRD) and among the most common microvascular complications of diabetes.6–9 Glomerular hypertrophy, tubulointerstitial inflammation, fibrosis, and glomerulosclerosis are pathological conditions caused by metabolic alterations associated with diabetes.10–15
DKD has a complicated and multifaceted pathogenesis, involving a combination of oxidative stress, the renin-angiotensin-aldosterone system (RAAS), the immune system, and inflammation.16 The conventional view is that DKD is a non-immune, metabolic, or hemodynamic glomerular disease caused by hyperglycemia, however research, has discovered that proinflammatory cytokine release and low-grade inflammation, both systemic and local, which are mainly due to inflammation driven by the innate immune system, are linked to DKD onset and progression.17 Abnormal metabolic states such as hyperglycemia and hyperinsulinemia lead to damage to the glomerular filtration membrane and apoptosis of renal tubular epithelial cells (TECs), and these passive effects elicit a response from the immune system.18–21 Immune cells recognize damage to kidney cells and release various cytokines to further promote the inflammatory response, driving kidney structure remodeling and interstitial fibrosis.17,22 Macrophages are a major class of innate immune cells, while T cells are important for adaptive immunity. Numerous experimental models of DKD and clinical trials have revealed the infiltration and activation of T lymphocytes and macrophages in the kidney.23–27
The number of individuals with ESRD resulting from DKD is rising annually, despite notable therapeutic advances,28 these patients require dialysis and kidney transplantation. The immune system contributes significantly to DKD; however, more research is needed to determine the exact mechanism by which immunity mediates the condition.
Role of the Immune System in DKD
The immune system comprises innate and adaptive arms. There are various classes of pattern recognition receptors (PRRs) in the innate immune system, such as nucleotide-binding NOD-like receptors (NLRs) and membrane-bound Toll-like receptors (TLRs), which recognize pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) in both extracellular and intracellular contexts.17 Unlike the adaptive immune system, the innate immune system recognizes endogenous danger signals by recognizing DAMPs, in addition to microbial byproducts, and can identify and eradicate the majority of microorganism-caused damage in a matter of minutes or hours.29 Rapid-acting cells of the innate immune response comprise granulocytes, mast cells, macrophages, and dendritic cells (DCs). Conversely, specificity, diversity, and memory are critical characteristics of the adaptive immune response, which differentiates self from non-self through specialized defense mechanisms involving specific recognition molecules.30 The primary cellular components of the adaptive immune system are T and B cells.
The innate immune system has a vital role in DKD pathogenesis. Prolonged exposure to high levels of blood glucose and advanced glycation end products (AGEs) results in injury or death of renal cells, leading to the release of DAMPs into the extracellular space. This danger signal is recognized by PRRs like TLRs (TLR2 and TLR4), NLRs, protease-activated receptors in the kinin-kallikrein system, the complement cascade, and other cell surface receptors. Activating these receptors triggers an inflammatory response in kidney-associated cells, while kidney injury leads to entry of monocytes from the bone marrow into the circulation, which differentiate monocytes into inflammatory macrophages and an increase in adhesion molecule expression on vascular endothelial cells. Further, the process of macrophage infiltration into damaged kidneys amplifies the inflammatory response. As signaling pathways, including transforming growth factor-β (TGF-β), mitogen-activated protein kinase (MAPK), and nuclear factor kappa-B (NF-κB), are activated, chronic and unresolved nephritis develops into progressive renal fibrosis31 (Figure 1). The complement system is crucial in innate immune defense via interactions with PRRs and drives inflammation. Glycosylation-induced inactivation of CD59 and hyperglycemia-induced activation of complement signaling combine to increase membrane attack complex deposition in tissues, which, in turn, activates intracellular signaling pathways leading to the release of proinflammatory cytokines and growth factors.32 There is growing evidence that the complement system contributes to DKD development. Additionally, complement activation can be used to identify patients at risk of this complication and serves as a target for therapy.33
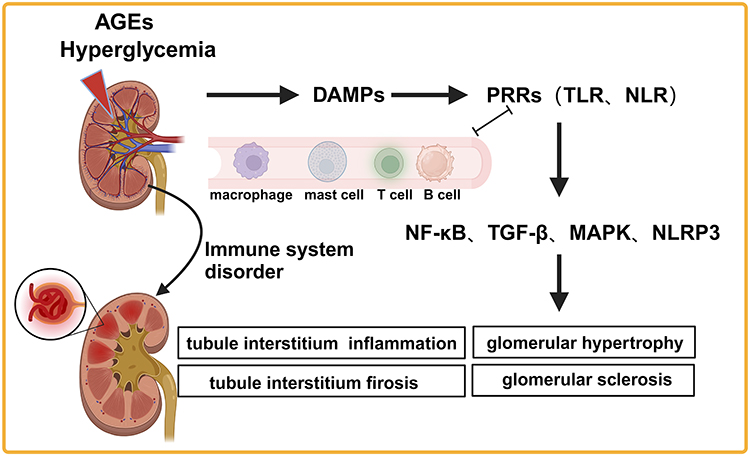 |
Figure 1 Immune cells are involved in DKD development. Abbreviations: AGEs, advanced glycation end products; DAMPs, danger-associated molecular patterns; PRRs, pattern recognition receptors; TLR, Toll-like receptors; NLR, NOD-like receptors; NF-κB, nuclear factor kappa-B; TGF-β, transforming growth factor-β; MAPK, mitogen-activated protein kinase; NLRP3, nucleotide-binding domain, leucine-rich repeat, and pyrin domain-containing protein 3. |
The adaptive immune system mainly functions through T and B cell activity. The kidneys of patients with type 2 diabetes mellitus (T2DM) have elevated activated T cell infiltration relative to those of nondiabetic patients, which is correlated with the degree of proteinuria in these patients.34 Further, increases in the percentage of Th17 cells and decreases in regulatory T cell (Treg) levels have been discovered in the blood of patients with type 1 diabetes; this disruption of the Treg/Th17 balance may exacerbate the development of microvascular complications of diabetes.35 Further, there is evidence implying that synergistic interaction of the adaptive immune system with specific inflammatory cytokines may be a fundamental element in the development of diabetic kidney damage.23 Additional research into the interactions between T cells and other immune cells in DKD, as well as examination of the differentiation and function of different T cell subpopulations in the context of dysregulated immune homeostasis, is necessary. Such research will be crucial for improving disease prediction and treatment.
Role of Immune Cells in DKD
Macrophage
Macrophages are major immune cells that infiltrate the kidneys in DKD, and their recruitment and activation promote the development of sclerosis and renal injury in individuals with diabetes.36 Further, activation of infiltrating and resident macrophages in the diabetic kidney leads to renal fibrosis and inflammation in the glomerular and tubulointerstitial compartments.37 Resident macrophages, which act as gatekeepers in the activation or inhibition of immunological responses, are rapidly activated by stimulation, and contribute significantly to DKD by producing cytokines and chemokines, attracting peripheral monocytes and macrophages, intensifying renal-associated cellular damage, and transforming into macrophage-myofibroblasts.38
Macrophages can be broadly categorized into M1 and M2 types according to their function and activation state. In the initial phases of renal damage, proinflammatory cytokines, DAMPs, PAMPs, and interferon gamma (IFN-γ) activate macrophages, initiating their transformation into a cells with an M1 phenotype, which are proinflammatory and react to cell damage.39 M1 macrophages contribute to inflammation by producing inducible nitric oxide synthase, as well as enhancing tissue inflammation and injury via the secretion of proinflammatory cytokines. Conversely, M2 macrophages reduce inflammation and promote fibrosis and wound repair.40 The M1/M2 macrophage ratio varies dynamically as DKD progresses through different phases. Renal biopsies revealed that, in the early stages of DKD (stages I and IIa), M1 macrophages are recruited to the kidney. The M1/M2 macrophage ratio peaks in the early stage of DKD and drops to a minimum later in the disease (stage III), when M2 macrophages become dominant.41 In renal samples from patients with DKD, glomerular CD163+ (an M2/anti-inflammatory marker) macrophages are positively correlated with DKD grade, interstitial fibrosis, tubular atrophy, and glomerulosclerosis.36
The regulation of macrophage recruitment and activation in DKD is a complex process involving various mechanisms. High glucose levels promote M1 macrophage transformation and podocyte apoptosis while reducing Sirt6 expression. Conversely, the overexpression of Sirt6 enables podocyte protection through M2 macrophage activation.42 Factors such as hyperglycemia, oxidative stress, and DAMPs promote the activation of the nucleotide-binding domain, leucine-rich repeat, and pyrin domain-containing protein 3 (NLRP3) inflammasome in macrophages, leading to the production and release of inflammatory cytokines, such as Interleukin-1 Beta (IL-1β) and Interleukin-18 (IL-18), which exacerbate the inflammatory response. NLRP3 knockout (KO) mice exhibit improved renal function, reduced glomerular hypertrophy, glomerulosclerosis, and inflammation under diabetic conditions.43 CAY10603, as a specific inhibitor of HDAC6 (histone deacetylase 6), significantly reduces pyroptosis in macrophages by suppressing the activation of the NLRP3 inflammasome, thereby demonstrating its therapeutic potential in the treatment of DKD.44 Bruton’s Tyrosine Kinase (BTK), an essential factor in immunomodulation, is activated in the kidneys of patients with DKD, and this activation correlates with proteinuria, creatinine levels, estimated glomerular filtration rate (eGFR), and progression of renal pathological changes.45 BTK KO in a mouse model has been shown to alleviate diabetic kidney damage, characterized by reduced urinary protein and decreased kidney inflammation, which is associated with reduced activation of the NLRP3 inflammasome in macrophages.45 A BTK inhibitor (PCI-32765) blocks macrophage-generated MAPK and NF-κB pathways, countering the pro-inflammatory consequences of high glucose,46 suggesting that it has potential as an immunosuppressant against DKD-induced inflammation, particularly hyperglycemia-induced responses. Variants of the gene encoding tonicity-responsive enhancer-binding protein (TonEBP) are robustly associated with inflammation and renal function in healthy populations. Furthermore, TonEBP mediates pro-inflammatory activation and macrophage migration induced by hyperglycemia in a DKD mouse model, leading to increased inflammation and renal injury.47
There is significant deposition of ApoC1 in the glomeruli of kidneys from patients with DKD, and ApoC1 overexpression can increase TNF-α and TGF-β cytokine-induced glomerulosclerosis in macrophages.48 Glycosphingolipid expression is also increased in DKD kidneys, which mediates inflammatory responses by enhancing TLR4 in human monocytes and mouse bone marrow-derived macrophages.49 Serum mannose-binding lectin (MBL) is a complement-activated molecule that recognizes carbohydrates and is associated with DKD. MBL triggers inflammation in macrophages by activating the NF-κB pathway and promotes macrophage, conversion to the M1 phenotype, independently of the complement lectin pathway.50 P2X7R is among multiple adenosine-5’-triphosphate-activated receptors; is expressed in macrophages, mesangial cells, and other cell types; and regulates MCP-1 secretion in cell models. Renal biopsy samples from patients with DKD exhibit high P2X7R expression levels, and mesangial P2X7R activation can stimulate early glomerular macrophage recruitment and prioritize activation of M1-polarized pro-inflammatory macrophages. A selective P2X7R inhibitor has potential to reduce renal macrophage accumulation, provide renal protection, and become a therapeutic agent in the future.51
Immune cells, particularly macrophages, and intrinsic renal cells interact closely in DKD to promote disease progression (Figure 2). In diabetes, the conditions of high glucose, AGEs, and albumin levels lead to increased heparinase expression in glomerular cells, where heparinase stimulates macrophage-mediated inflammation, leading to chronic kidney injury.52 Hence, heparinase is associated with macrophage activation, chronic inflammation, and kidney injury in diabetes. In DKD kidneys, IL-1β is mainly derived from renal TECs, which promote macrophage polarization towards a pro-inflammatory M1 phenotype and secretion of IL-6, leading to sodium retention and salt sensitivity.18 M1 macrophages accelerate cellular senescence by inducing elevated reactive oxygen species levels and p38 MAPK signaling activation in human renal glomerular endothelial cells.53 Plasma growth hormone (GH) levels are increased in individuals with type 1 diabetes, and GH-stimulated podocytes have the potential to cause macrophage accumulation by secreting TNF-α and prompting macrophage migration, potentially serving as an independent factor mediating DKD progression in patients with type 1 diabetes.54 In diabetic mice, a reduction in angiotensin II type 1 receptor-associated protein within the renal tubules results in hyperactivation of the tubular angiotensin II type 1 receptor signaling/RAAS system, while CD206 expression, which is considered a marker of M2 macrophages, is decreased in the renal tubular interstitium. Tubulo-glomerular crosstalk leads to increased levels of TNF-α and oxidative stress components in the glomerulus, as well as exacerbation of tubular hypertrophy and podocyte injury.55 T-cell immunoglobulin structural domain and mucin structural domain-3 (Tim-3) are a negative regulator of Th1 immunity and have complex roles in regulating macrophage activation. Increased Tim-3 expression in renal macrophages through activation of the NF-κB/TNF-α pathway was found in patients with DKD and diabetic mice, as well as damaged podocytes.56 The knockout of angiopoietin-like protein 3 alleviates podocyte epithelial–mesenchymal transition and injury in DKD by promoting the transformation of M1 to M2 macrophages, inhibiting NLRP3 inflammasome activation, and suppressing IL-1β release.57
 |
Figure 2 Interactions of immune and renal cells. Abbreviations: Tim-3, T-cell immunoglobulin structural domain and mucin structural domain-3; TNF-α, tumor necrosis factor-alpha; Sirt6, sirtuin 6; Atg9b, autophagy-related protein 9B; A20, tumor necrosis factor alpha-induced protein 3; DR5, death receptor 5; IL-6, interleukin-6; HIF-1α, hypoxia-inducible factor 1-alpha; IL-17, interleukin-17; IFN-γ, interferon gamma; NETs, neutrophil extracellular traps; TGF-β, transforming growth factor-β. |
Non-coding RNAs and exosomes contribute to the interactions between kidney cells and macrophages in DKD. Elevated concentrations of miR-19b-3p in urine exosomes are correlated with tubulointerstitial inflammation in patients with DKD. Furthermore, damaged renal TECs communicate with macrophages through the exosome/miR-19b-3p/SOCS1 axis, leading to M1 macrophages activation and exacerbation of tubulointerstitial inflammation.58 Macrophage infiltration around lipotoxic renal TECs has also been detected in patients with DKD, and lipotoxic TEC-derived extracellular vesicles (EV) activate the inflammatory phenotype of macrophages and induce production of macrophage-derived EV, which can induce TEC apoptosis by a death receptor 5 (DR5)-dependent process.19 Furthermore, miR-21-5p in macrophage-derived EV can regulate A20 and cause podocyte injury.59 High levels of proteinuria can stimulate the expression of inflammatory and fibrotic genes in DKD, by increasing HIF-1α-induced glycolysis in renal macrophages, through enhancement of EV production by renal TECs.60 Macrophage-derived exosomes stimulated by high glucose (HG-exo) cause renal mesangial proliferation and extracellular matrix (ECM) accumulation. HG-exo induces dysfunction and inflammation in mouse TECs by carrying excess miR-7002-5p and inhibits autophagy by targeting Atg9b.61 Glomerular mesangial cells (GMCs) are involved in DKD development by promoting NLRP3 inflammasome activation and autophagy defects in response to high glucose-treated macrophage-derived exosomes.62 Furthermore, Epsin1 mediates the activation of exosome sorting induced by Dll4 and Notch1, to regulate tubular macrophage crosstalk in DKD.63 In contrast, miR-93-5p and TLR4 expression are upregulated by exosomes released by M2 macrophages, which reduces LPS-induced podocyte apoptosis.64 M2 macrophages also ameliorate high-glucose-mediated podocyte injury by suppressing DUSP1 expression, secreting exosomes containing miR-25-3p, and activating cellular autophagy.65
T Cells
T lymphocytes are classified into CD4 and CD8 subpopulations, based on cell surface markers, and have vital functions in pathogen removal and host defense. Based on their cytokine profiles and functional roles, CD4+ T cells can be further subdivided into T helper (Th) cells and Tregs. Antigens in the context of class I major histocompatibility (MHC-I) molecules are specifically recognized by CD8+ T cells, which mainly function as cytotoxic T cells. CD8+ and CD4+ T cells are defined as conventional T cells; however, unconventional T cells, that respond rapidly upon encountering antigen, are also present in the kidney, including mucosa-associated invariant T, natural killer T and γδ T cells.66 Additionally, a class of T cells known to reside in the kidney has been identified as tissue-resident memory (TRM) T cells, which are the most prevalent subpopulation of memory T cells.67 These different T cell subtypes regulate one another in various ways and work together to maintain immune homeostasis in the kidney.
T cells, and their activation markers, are actively expressed in the periphery and kidney in DKD. Kidneys from diabetic mice have significantly increased levels of CD3+ T cells, which generate TNF-α and IFN-γ. BTBR ob/ob mice have a phenotype similar to advanced human DKD, with increased infiltration of T lymphocytes (CD3+, CD 4+, and γδ lymphocytes) and increased expression of Th17 drivers, such as IL-6, observed in their kidneys.28 Furthermore, patients with type 2 diabetes have significantly higher levels of CD4+, CD8+, and CD20+ cells in the renal interstitium, and their proteinuria levels are correlated with CD4+ and CD20+ cell numbers.34 Furthermore, the numbers of IFN-γ+CD4+ and IL-17A+CD4+ T cells in the kidneys increase significantly from the onset of albuminuria, and the common immunosuppressant, mycophenolate mofetil, can suppress intrarenal IL-17A + CD4+ T cells from early DKD and ameliorate proteinuria.27 KIM-1 and CD8+ T cell infiltrates were detected in renal biopsies from patients with DKD. Glycemic changes increase KIM-1 production from CD8+ T cells, thus raising the risk of DKD in patients with diabetes.68 Increased numbers of peripheral blood CD4+CR5+PD-1+ THF cells were also found in patients with DKD,69 and patients with type 2 diabetes and proteinuria express more CD25 and CD69 on their circulating CD8+ T cells.70 Moreover, the serum levels of T-lymphocyte activation markers, specifically CD28 and CTLA-4, increase as DKD progresses. Low eGFR levels and high urinary TNF-α are positively correlated with CTLA-4, indicating that activation of the TNF-α pathway and T-lymphocyte-mediated immune responses are important in the early stages of DKD.71 In patients with DKD, there is a relationship between renal CD3+ T cell accumulation and urinary CCL21 levels, which are derived from small EV and co-localize with CCL21 in the tubular interstitium. CCL21-mediated T cell infiltration may be an important chronic inflammation pathway in DKD.72 Analysis of peripheral blood samples from patients, with early DKD using the mass cell counting technique, CyTOF, revealed a significant decrease in B cells, a significant increase in monocytes, and a notable increase in Th1 cells and Treg.73
Several studies have reported a correlation between elevations of T cell counts and levels of their activation markers with DKD and indicators, such as renal function, suggesting that T cell activation is a significant factor in DKD. Studies on renal fibrosis have demonstrated that T cells stimulate renal TECs to produce specific chemokines, such as CCL2 and CXCL10, through the release of pro-inflammatory factors, including TNF-α and IL-17, thus recruiting more T cells and other immune effectors to the kidney and inducing renal TECs to transform into mesenchymal fibroblast-like cells. Further, by releasing fibroblastogenic factors, such as TGF-β, immune effectors activate mesenchymal fibroblasts and promote their proliferation and secretion of stromal matrix.74 Among T cells, Th cells primarily promote or inhibit inflammatory responses by secreting cytokines, including TNF-α, IFN-γ, IL-17, IL-10, and TGF-β, which impact renal inflammation, repair, and fibrosis.75 It is currently thought that, in DKD, T cells are either expanded, differentiated, and activated in the kidney, or are recruited from the circulation to renal tissues. These cells can have either harmful or beneficial effects through various mechanisms, including modifying insulin resistance, injuring podocytes, encouraging fibrosis, and controlling proteinuria. Th1, Th2, Th17, Treg, and cytotoxic T cells have all been implicated in the development of DKD.76
Investigation of the roles of T cells in DKD has become the focus of significant attention, and some potential mechanisms by which T cells promote DKD and targets for T cell regulation have recently been discovered. Biglycan is an ECM proteoglycan that is upregulated at all stages of DKD, and stimulates CXCL10 and CCL20 production by macrophages through the biglycan/TLR/TRIF/myd88 signaling pathway, thereby recruiting Th1 and Th17 cells. Further, IFN-γ can synergistically stimulate macrophages to enhance CXCL9 expression and attract CXCR+ Th1 and Th17 cells, enhancing the infiltration of T cell subsets.77 In addition, IFN-γ can induce GMCs to express antigen-presenting and co-stimulatory molecules, such as MHC-II, ICAM-1, CD40, and CD80, which act as non-professional antigen-presenting cells, while uptake and presentation of antigenic peptides by activated GMCs induces CD4+ T cell (Th0 cells) polarization toward a Th1 phenotype and promotes inflammatory responses.78 Liang Li et al79 found that the percentage of CD8+ TRM cells was significantly increased in kidneys from DKD mice and patients. IL-15, which is highly expressed in DKD kidneys, significantly promotes the development and activation of CD8+ TRM cells, thereby inducing podocyte injury and glomerulosclerosis. Sparsentan can regulate renal CD8+ TRM cells by blocking angiotensin II endothelin-1 mediated IL-15 signaling. In kidney biopsy specimens from patients with type 2 diabetes, the number of CD4+IL-17+ T cells is positively associated with eGFR deterioration, and IL-6 may be synergistically enhanced by IL-17 and CD40L. Further, production of MCP-1, TGF-β, and NF-κB mediates the inflammatory response and remodeling linked with tissue damage and glomerulosclerosis in DKD.80
Mast Cells
Mast cells are pluripotent cells derived from bone marrow that contain abundant mediators of inflammation and growth factors. Mast cell precursors are hematopoietic progenitor cells that migrate to injured tissue from the bone marrow and mature into local mast cells. Resident renal mast cells may contribute to DKD development by releasing various pathogenic substances, including chymase, TGF-β, cathepsin G, tryptase, renin, histamine, and proinflammatory cytokines.81 Further research is required to validate the potential role of these mast cell-derived mediators in the intricate pathophysiology of DKD.
Immunohistochemical staining of renal tissues from patients with DKD of varying clinical stages revealed increases in the degree of degranulation and quantity of mast cells as the disease progressed. Mast cells may induce renal inflammation and fibrosis by degranulating and releasing bioactive chemicals into the tubular interstitium, including chymase, tryptase, TGF-β1, renin, and TNF-α.82 Rats with DKD kidneys also showed an increase in mast cell counts, while application of the renin–angiotensin system blocker, aliskiren (AL), mitigated the rise in mast cell numbers, indicating a potential protective effect of AL on the kidney,83 however, the exact mechanism involved requires further investigation. Mast cell infiltration may cause renal interstitial fibrosis through the SCF/c-kit signaling pathway in a rat model of DKD, while tranilast, an anti-allergic medication, can prevent renal interstitial fibrosis by reducing mast cell infiltration in this context.84 SERPINA3, an inhibitor of mast cell chymase, is upregulated in the renal tubules of patients with DKD, and may serve as a protective immune-associated molecule that can prevent renal interstitial fibrosis by inhibiting mast cell activation and multiplication, as well as downregulating chymotrypsin activity, to prevent renal tubular injury.85
B Cells
B lymphocytes are derived from pluripotent stem cells in the bone marrow, which transform into plasma cells in response to antigenic stimulation. Plasma cells produce and release antibodies, thereby contributing to humoral immunity.86
B-cell infiltration and immune complex deposition have been observed in kidneys affected by DKD. A study conducted on children with type 1 diabetes mellitus demonstrated a relationship between elevated levels of circulating IgG immune complex and the development of early DKD.87 In addition, increased deposition of IgG+ B cells was detected in the glomeruli of diabetic NOD mice.88 Furthermore, immune complexes containing oxidized low-density lipoprotein cholesterol are thought to promote mesangial expansion in the glomeruli by stimulating type IV collagen production.89 Among B cell subsets, CD38+CD19+ and CD38+CD19+CD40+ B cells were significantly elevated in peripheral blood from both patients with DKD and healthy controls, while CD38+CD19+ B cells number was positively correlated with serum IgG levels and albumin excretion rate and negatively correlated with eGFR.69 Consequently, B cells are thought to contribute to DKD by generating antibodies and building immune complexes that are deposited in the kidney.
Immunological complexes in the glomerulus trigger complement activation and cytokine release, which in turn drive macrophage aggregation and inflammation. The release of DAMPs after ECM injury in DKD may activate B cells.90 Regulatory B cells (Bregs), a unique B cell subpopulation, downregulate immune responses, and their absence is linked to increased autoimmune responses. Bregs cells secrete IL-10 to maintain immune tolerance in the body. Numbers of circulating CD19+CD24hiCD38hi Bregs are reduced in patients with DKD. Further, the number of circulating CD19+CD24hiCD38hi B cells in patients with DKD is positively correlated with eGFR and serum IL-10 levels, but negatively correlated with urinary protein levels,91 implying that DKD development also involves Bregs. The potential beneficial effects of Bregs represent a novel avenue for exploration in the context of DKD treatment; however, studies on B cells in DKD are scarce and further exploration is necessary.
Neutrophils
Neutrophils are a subtype of granulocytes and the most abundant type of white blood cells with important functions in the immune system. These phagocytes have short life spans and are quickly mobilized to sites of infection or tissue injury, however, their involvement in DKD remains unclear. The neutrophil–lymphocyte ratio (NLR) is higher in patients with diabetes and nephropathy, and NLR is positively correlated with C-reactive protein levels and microalbuminuria, but negatively correlated with eGFR.92,93 The NLR is also used to evaluate the degree of neutrophil-related chronic inflammation (NRCI); however, a randomized, double-blind, controlled clinical trial discovered that, while low-dose colchicine therapy (0.5 mg/day) was effective and safe in reducing NRCI, it did not delay the onset of DKD.94
Activation of neutrophils causes DNA decondensation and histone deamination, particularly of peptidylarginine deiminase 4 (PAD4), which catalyzes histone citrullination.95 Subsequently, neutrophil extracellular traps (NETs), which are composed of histones, DNA, and neutrophil proteases, such as neutrophil elastase (NE) and myeloperoxidase, are released.96 Increased deposition of NETs has been identified in the glomeruli of patients and mice with DKD.97,98 NETs induce dysregulation of several genes related to cellular membrane function, resulting in pore formation in cell membranes and pyroptosis in glomerular endothelial cells.97 Under high glucose conditions, NETs can enhance glomerular endothelial dysfunction and NLRP3 inflammasome activation both in vivo and in vitro, whereas inhibition of NETs (eg, with PAD4 inhibitors) can alleviate endothelial dysfunction and renal damage in DKD.98 In T2DM, inflammation and neutrophil activation promote NE release, which is mediated by regulation of the endogenous inhibitors, alpha1-antitrypsin (α1-AT) and alpha2-macroglobulin (α2-MG), under normal physiological conditions. NE activity levels are significantly elevated in the plasma of patients with DKD, while α1-AT and α2-MG levels are decreased.99 In summary, therapy targeting reduction of NETs secretion and maintenance of NE homeostasis may be promising for halting DKD progression.
Group 2 Innate Lymphoid Cells (ILC2s)
ILC2s are innate immune cells that respond rapidly to their surroundings via soluble inflammatory mediators and intercellular interaction100 and play a critical role in Th1/Th2 homeostasis.101 ILC2s contribute to tissue healing, allergic inflammation, metabolic balance, and tissue fibrosis, including pulmonary fibrosis, intestinal fibrosis, and hepatic fibrosis.102,103
Although less studied, ILC2s may also impact the course of DKD. In patients with DKD, mRNA levels of factors encoding the ILC2 associated molecules, ROR, T1/ST2, and IL-5/IL-13, are increased. Further, IL-13 and T1/ST2 mRNA expression levels are associated with fasting glucose, lipids, and body weight,104 indicating that ILC2s may contribute to DKD by regulating blood pressure, lipid metabolism, and other factors that contribute to metabolic syndrome. Cuiping Liu et al105 discovered that the proportions of ILC2 and Th2-associated cytokines, IL-4, IL-5 and IL-13, are higher in peripheral blood from patients with DKD, and positively associated with DKD severity. Furthermore, when HK-2 cells were stimulated with IL-4 or IL-13, TGF-1 and fibronectin (FN) production increased, indicating that ILC2s may have a role in renal fibrosis in DKD via the TGF-1 signaling pathway.
Single-Cell Analysis of Immune Cells in DKD
The development of single-cell RNA sequencing (scRNA-seq) techniques in recent years has increased the recognition of the roles of immune cells in renal disease.106 Unlike bulk RNA sequencing, scRNA-seq can identify transcription status and signaling pathways in many cell types while quantifying gene expression in a single cell. A research project using both scRNA-seq and bulk RNA-seq revealed that the recruitment and activation of M1 macrophages and cytotoxic T cells initiates inflammation, which is a major contributor to permanent kidney damage.107 Recent developments in library preparation and separation techniques, such as single nucleus RNA sequencing (snRNA-seq), have facilitated discovery of unusual cell types in cryopreserved samples.108 Relative to the control group, snRNA-seq of renal biopsy specimens from individuals with type 2 diabetes and early-stage DKD showed a 7- to 8-fold increase in leukocytes. Additionally, increased expression levels of TNFRSF21 in infiltrating CD14+ monocyte subpopulations, IL-R1 in CD16+ monocytes and antigen-presenting cells, and IL-18R1 in CD4+ and CD8+ T cells were detected.109 ScRNA-seq of glomeruli in diabetic mice identified a higher number of immune cells, where macrophages were predominant, with more cells expressing M1 than M2 phenotypic markers.110 Fu et al111 conducted scRNA-seq on CD45-enriched kidney immune cells from DKD mice and focused on analysis of mononuclear phagocytes. They found that, as DKD progressed, resident and infiltrating macrophage subpopulations in the kidneys increased, accompanied by subpopulation-specific increases in pro- or anti-inflammatory gene expression and gene expression tending towards an undifferentiated phenotype, while M1-like inflammatory phenotypes increased over time. Furthermore, based on scRNA-seq data from db/db mice kidneys, it has been predicted that M1 macrophages can target renal stromal cells via SPP1 and activate T cells via the MHC-II pathway, which then recognize other renal cells via the MHC-I pathway;112 however, this study by Fu et al raises several unanswered questions.113 Renal inflammation is a result of intricate cell–cell communication among various cell types. The interaction between the identified subpopulation of activated macrophages and the renal parenchyma remains unclear, due to loss of spatial information during cell dissociation. The evolving field of transcriptomics, which has spatial resolution capabilities, may provide answers to these questions. Second, the regulation of macrophage activation and polarization at the genomic level in DKD remains largely unexplored. Understanding of gene regulatory networks necessitates thorough analysis of multi-group single-cell datasets comprising transcriptomes and chromatin accessibility within the same cell. Furthermore, a comprehensive investigation into immune cell heterogeneity in human DKD is imperative, to augment findings garnered from preclinical mouse models.
Regulation of gene expression involves a dynamic interplay between control of chromatin structure and accessibility, and recruitment of transcription factors to promoter regions, enhancers, and activator sequences.114 Accessible chromatin regions can be identified using DNase-seq or assay for transposase accessible chromatin with high-throughput sequencing (ATAC-seq).115 Single-cell ATAC-seq studies of epigenomic landscapes promise to reveal heterogeneity among cells in gene regulatory programs;116 nevertheless, there has been a lack of ATAC-seq studies in the context of DKD.
ScRNA-seq and ATAC-seq datasets are generated post-tissue isolation, leading to loss of spatial and morphological details about cells in the tissue environment. This knowledge is vital to understand the role of various cells in tissue function, particularly for organs with intricate spatial structures, such as the kidney. The progressive development of spatial transcriptomics may address these issues, but no high-throughput spatial transcriptomics studies on the kidney have yet been published. In the future, new insights into renal homeostasis and disease are expected to be provided by spatial transcriptomics.
Additionally, the field of single-cell proteomics is developing rapidly. Single-cell proteomics technologies, including CITE-Seq (Cellular Indexing of Transcriptomes and Epitopes by Sequencing), have recently emerged, and can be combined with sequencing-based methods to simultaneously measure proteins and transcripts at single-cell resolution.117 It is anticipated that these technologies will gain wider use in kidney research in the near future, to provide information complementary to other single-cell datasets.
Bioinformatics Analysis of Immune Cells in DKD
Thanks to advances in bioinformatics, efficient algorithms have accelerated the transformation of all types of omics big data into novel therapeutic targets. CIBERSORT is a robust method for assessing immune cell infiltration and is increasingly used in conjunction with bioinformatics approaches to develop novel diagnostic markers and research immune cell infiltration patterns in tissues. By using CIBERSORT to identify immune cells, infiltration of M1 and M2 macrophages, memory B cells, γδ T cells, and resting mast cells in DKD glomeruli were found to be increased in DKD glomeruli, while neutrophil and activated mast cell infiltration were relatively reduced,118–121 however, the infiltration and state of immune cells, such as NK cells, Tregs and DCs appears to be unstable in DKD glomerular tissues. Yan Jia et al122 found an up-regulation of M1 macrophages, CD4+ T cells, CD8+ T cells, Th2 cells, conventional DCs, and activated DCs, but a down-regulation of Tregs in DKD renal tubulointerstitial tissue. Furthermore, increased expression of VCAM1 in renal tubules and interstitial immune cell infiltration contribute to DKD development. Zheng Ye et al123 analyzed high-throughput RNA-SEQ data (GSE142025) from early and late-stage DKD and discovered that immune spectrum diversity and abundance were significantly increased in patients with late-stage DKD, while immune function in patients with early-stage DKD was not significantly changed. In contrast, immune-related signaling pathways, primarily concentrated in immune cells are significantly activated in early-stage DKD. B cell-mediated antibody responses may play an essential part in DKD pathogenesis.
Through analysis of online single-cell RNA profiles of patients with DKD (GSE131882), B cells, T cells, and plasma cells were identified as involved in DKD progression and may be contribute to crosstalk with TECs.124 Cell crosstalk analysis using CellPhoneDB based on GSE131882 suggested that CD20 expression was significantly increased in the DKD group.125 Drugs that target antibodies against CD20 or B cells hold promise as potential DKD therapeutic options. As previously reported, the mTOR pathway contributes to DKD pathogenesis.126 By analyzing GSE131882, Xi Lu et al also found that the differentially expressed immune cell marker genes (RICTOR, PRKCB, and EIF4B) were significantly enriched in the mTOR pathway and confirmed these results using RT-qPCR and Western blot experiments in DKD rat models.127
Bioinformatics analysis has also provided more clues to the interactions between immune cells and DKD. Skewed gene patterns in both particular immune cells in the bloodstream and equivalent immune cells infiltrated in DKD kidneys indicated significant enrichment of Fc epsilon receptor (FcER1) and T-cell receptor-mediated signaling. Elevated FcER1 gene and protein levels were observed in the kidneys of patients with DKD and were co-localized with infiltrating mast cells.128 FcER1 activation may lead to release of inflammation mediators from mast cells, thereby exacerbating DKD. Indoleamine 2,3-dioxygenase 1 (IDO1) was identified as a core immune gene with increased expression in mice with DKD. M1 macrophage and monocyte numbers were positively correlated while resting memory CD4+ T cell number was negatively correlated, with IDO1 upregulation. In vitro experiments verified that high glucose stimulation resulted in upregulation of IDO1 expression in peritoneal macrophages and that inhibition of IDO1 reversed the production of inflammatory factors.129 Qiannan Xu et al130 identified VCAN as a hub gene related to immune injury in DKD tubulointerstitial injury, levels of its translated protein, versican, in patient renal tissues are associated with immune cell accumulation in the kidneys. CCL19 levels were increased in DKD kidneys and HK-2 cells treated with high glucose, where CCL19 is considered a core molecule in DKD, able to regulate lymphocyte circulation, as well as B- and T-cell migration in the thymus and secondary lymphoid organs, through binding to the CCR7 receptor.131 Xiaohui Li et al132 identified CASP1, MS4A4A, CD53, and GBP2 as hub genes implicated in macrophage-mediated inflammation in DKD. Further experiments showed that GBP2 can promote DKD progression by activating pro-inflammatory M1 macrophages via the notch1 signaling pathway.
Meng Zhou et al133 discovered that various signaling pathways, such as those involving VISTANT, SPP1, and IGF, as well as receptor-ligand pairs, including NRG1-ERBB4, SPP1-CD44, Igf1-Igf1r, and NAMPT-INSR, may play significant roles in DKD by inducing crosstalk between renal and immune cells. Hypoxia-related immune molecules, including PSMB8, PSMB9, RHOA, VCAM1, and CDKN1B, may regulate T cells after tubular lesions, whereas APOID1, TGFBR3, KDR, and CPEB1 may influence T cells after glomerular injury.134 LCK and HCK were identified as two core genes, levels of which were increased in DKD. Correlation analysis revealed that both were associated with active DC, CD8+ T cells, CD4+ T effector memory, CD8+ T effector memory, and mast cells.135 Xueqin Zhang et al136 identified SLIT3, PDE1A, and CFH as hub genes closely associated with DKD, which are significantly positively correlated with γδ T cell, M2 macrophage, and resting mast cell levels. Mingming Xu et al137 identified three immunological and oxidative stress-related hub genes (CD36, SLC1A3, and ITGB2) by merging weighted gene co-expression network analysis (WGCNA), protein–protein interaction (PPI) networks, and machine learning data; however, because their results were based solely on bioinformatics analysis, with no basic experiments to validate them, it is unclear whether the conclusions are truly valid.
Biomarkers linked to immune infiltration provide a fresh perspective to inform future investigations of DKD diagnosis and therapy options. Jing Chen et al138 identified six pro-inflammatory and pro-fibrotic candidate targets (including CCR2, MOXD1, COL6A3, COL1A2, PYCARD, and C7) as potential DKD susceptibility genes and validated them by RT-qPCR in kidney samples from DKD mice. Iron concentration changes are intimately related to the variety and complexity of the immunological milieu in patients with DKD, and The Iron Death Scoring System can establish links between iron content and immune cell infiltration. To this end, JingYuan Ma et al139 identified the iron death core genes (PRDX6, DUSP1, PEBP1, GABARAPL1, ZFP36, RGS4, and TSC22D3) as key factors in iron concentration and immune inflammation, and these genes reliably distinguished DKD from control samples. Wang Yuejun et al140 used VEGFC, FN1, and complement component 3 as immunological biomarkers of DKD, and found that they were positively correlated with M2 macrophage levels. Clinical database verification demonstrated that, VEGFC is a significantly correlated with eGFR in patients with DKD. Juan Jin et al73 identified biomarkers, including CTLA-4, CXCR3, CCR4, CD39, PD-1, and HLA-DR, via mass cytometry (CyTOF) analysis, which are associated with significantly altered monocyte, T cell, and B cell subpopulations in early-stage DKD; however, further validation in other cohorts is necessary. Shaojie Fu et al121 used three different algorithms, including least absolute shrinkage and selection operator (LASSO), support vector machine-recursive feature elimination (SVM-RFE), and random forest (RF), to identify DUSP1 and PRKAR2B as possible biomarkers. Further, immunohistochemical staining indicated that DUSP1 and PRKAR2B expression levels were significantly decreased in patients with DKD. Moreover, expression of these two genes was negatively correlated with M2 macrophage infiltration and positively correlated with neutrophil infiltration. By applying the Venn diagram and machine learning algorithms (LASSO, RF, and SVM-RFE), Han et al141 found that PRKAR2B and TGFBI are potential diagnostic biomarkers for glomerular injury related to immune cell infiltration. These algorithms were also used to demonstrate that ADI1, PTGS2, DGKH, and POLR2B, which are associated with immune infiltration, have potential to serve as diagnostic biomarkers for DKD.142 Decreased expression levels of ADI1 and PTGS2 in patients with DKD are associated with enhanced immune system activation. The significance of DGKH and POLR2B in DKD requires further validation. Handi Zhou et al143 identified CCR2, CX3CR1, and SELP as potential diagnostic biomarkers for DKD, which were associated with checkpoints, cytolytic activity, macrophages, MHC class I molecules, and paraneoplastic inflammation.
Although bioinformatics analyses have yielded various results, small sample sizes and database limitations mean that the findings often require validation using basic experimental methods.
Conclusion
In summary, immune cell imbalance can cause disorders of the immune-inflammatory microenvironment through multiple signaling pathways, resulting in permanent kidney damage and the development of DKD. Therefore, further understanding of the molecular mechanisms underlying immune cell function in the context DKD is of great importance for improving renal disease. In recent years, the development of single-cell sequencing and bioinformatics technologies, to assess gene expression patterns in individual cells in populations of different origins, have facilitated better understanding of the dynamic evolution of immune cells in the kidney and analysis of the complex underlying DKD at the cellular level, providing new approaches to clinical diagnosis and treatment.
Acknowledgments
We would like to thank the Metabolic Vascular Disease Key Laboratory of Sichuan Province.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The work was supported by grants from the National Natural Science Foundation of China (NO.U22A20286), Sichuan Science and Technology Program (NO.2023YFS0471), Luzhou-Southwest Medical University cooperation project (NO.2021LZXNYD-P02).
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Sun H, Saeedi P, Karuranga S, et al. IDF diabetes atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabet Res Clin Pract. 2022;183:109119. doi:10.1016/j.diabres.2021.109119
2. Federation ID. IDF Diabetes Atlas.
3. Akdağ M, Özçelik AB, Demir Y, Beydemir Ş. Design, synthesis, and aldose reductase inhibitory effect of some novel carboxylic acid derivatives bearing 2-substituted-6-aryloxo-pyridazinone moiety. J Mol Struct. 2022;1258:132675. doi:10.1016/j.molstruc.2022.132675
4. Sever B, Altıntop MD, Demir Y, et al. Identification of a new class of potent aldose reductase inhibitors: design, microwave-assisted synthesis, in vitro and in silico evaluation of 2-pyrazolines. Chem. Biol. Interact. 2021;345:109576. doi:10.1016/j.cbi.2021.109576
5. Koye DN, Shaw JE, Reid CM, Atkins RC, Reutens AT, Magliano DJ. Incidence of chronic kidney disease among people with diabetes: a systematic review of observational studies. Diabet Med. 2017;34(7):887–901. doi:10.1111/dme.13324
6. Selby NM, Taal MW. An updated overview of diabetic nephropathy: diagnosis, prognosis, treatment goals and latest guidelines. Diabetes Obes Metab. 2020;22(Suppl 1):3–15. doi:10.1111/dom.14007
7. Cheng HT, Xu X, Lim PS, Hung KY. Worldwide epidemiology of diabetes-related end-stage renal disease, 2000-2015. Diabetes Care. 2021;44(1):89–97. doi:10.2337/dc20-1913
8. Sever B, Altıntop MD, Demir Y, et al. An extensive research on aldose reductase inhibitory effects of new 4h-1,2,4-triazole derivatives. J Mol Struct. 2021;1224:129446. doi:10.1016/j.molstruc.2020.129446
9. Demir Y, Ceylan H, Türkeş C, Beydemir Ş. Molecular docking and inhibition studies of vulpinic, carnosic and usnic acids on polyol pathway enzymes. J Biomol Struct Dyn. 2022;40(22):12008–12021. doi:10.1080/07391102.2021.1967195
10. Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: challenges, progress, and possibilities. Clin J Am Soc Nephrol. 2017;12(12):2032–2045. doi:10.2215/CJN.11491116
11. DeFronzo RA, Reeves WB, Awad AS. Pathophysiology of diabetic kidney disease: impact of sglt2 inhibitors. Nat Rev Nephrol. 2021;17(5):319–334. doi:10.1038/s41581-021-00393-8
12. Tuttle KR, Agarwal R, Alpers CE, et al. Molecular mechanisms and therapeutic targets for diabetic kidney disease. Kidney Int. 2022;102(2):248–260. doi:10.1016/j.kint.2022.05.012
13. Gupta S, Dominguez M, Golestaneh L. Diabetic kidney disease: an update. Med Clin North Am. 2023;107(4):689–705. doi:10.1016/j.mcna.2023.03.004
14. Sever B, Altıntop MD, Demir Y, Akalın Çiftçi G, Beydemir Ş, Özdemir A. Design, synthesis, in vitro and in silico investigation of aldose reductase inhibitory effects of new thiazole-based compounds. Bioorg. Chem. 2020;102:104110. doi:10.1016/j.bioorg.2020.104110
15. Demir Y, Tokalı FS, Kalay E, et al. Synthesis and characterization of novel acyl hydrazones derived from vanillin as potential aldose reductase inhibitors. Molecular Diversity. 2023;27(4):1713–1733. doi:10.1007/s11030-022-10526-1
16. Samsu N. Diabetic nephropathy: challenges in pathogenesis, diagnosis, and treatment. Biomed Res Int. 2021;2021:1497449. doi:10.1155/2021/1497449
17. Wada J, Makino H. Innate immunity in diabetes and diabetic nephropathy. Nat Rev Nephrol. 2016;12(1):13–26. doi:10.1038/nrneph.2015.175
18. Veiras LC, Bernstein EA, Cao D, et al. Tubular il-1beta induces salt sensitivity in diabetes by activating renal macrophages. Circ Res. 2022;131:59–73. doi:10.1161/CIRCRESAHA.121.320239
19. Jiang WJ, Xu CT, Du CL, et al. Tubular epithelial cell-to-macrophage communication forms a negative feedback loop via extracellular vesicle transfer to promote renal inflammation and apoptosis in diabetic nephropathy. Theranostics. 2022;12:324–339. doi:10.7150/thno.63735
20. Tokalı FS, Demir Y, Türkeş C, Dinçer B, Beydemir Ş. Novel acetic acid derivatives containing quinazolin-4(3 H)-one ring: synthesis, in vitro, and in silico evaluation of potent aldose reductase inhibitors. Drug Dev. Res. 2023;84(2):275–295. doi:10.1002/ddr.22031
21. Ertano BY, Demir Y, Nural Y, Erdoğan O. Investigation of the effect of acylthiourea derivatives on diabetes-associated enzymes. ChemistrySelect. 2022;7(46):e202204149. doi:10.1002/slct.202204149
22. Zheng Z, Zheng F. Immune cells and inflammation in diabetic nephropathy. J Diabetes Res. 2016;2016:1841690. doi:10.1155/2016/1841690
23. Kong L, Andrikopoulos S, MacIsaac RJ, et al. Role of the adaptive immune system in diabetic kidney disease. J Diabetes Investig. 2022;13:213–226. doi:10.1111/jdi.13725
24. Chow F, Ozols E, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury. Kidney Int. 2004;65(1):116–128. doi:10.1111/j.1523-1755.2004.00367.x
25. Chow FY, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in streptozotocin-induced diabetic nephropathy: potential role in renal fibrosis. Nephrol Dial Transplant. 2004;19(12):2987–2996. doi:10.1093/ndt/gfh441
26. Ma T, Li X, Zhu Y, et al. Excessive activation of notch signaling in macrophages promote kidney inflammation, fibrosis, and necroptosis. Front Immunol. 2022;13:835879. doi:10.3389/fimmu.2022.835879
27. Kim SM, Lee SH, Lee A, et al. Targeting t helper 17 by mycophenolate mofetil attenuates diabetic nephropathy progression. Transl Res. 2015;166:375–383. doi:10.1016/j.trsl.2015.04.013
28. Lavoz C, Matus YS, Orejudo M, et al. Interleukin-17a blockade reduces albuminuria and kidney injury in an accelerated model of diabetic nephropathy. Kidney Int. 2019;95(6):1418–1432. doi:10.1016/j.kint.2018.12.031
29. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22(2):240–273. doi:10.1128/CMR.00046-08
30. McComb S, Thiriot A, Akache B, Krishnan L, Stark F. Introduction to the immune system. Methods Mol Biol. 2019;2024:1–24.
31. Tang SCW, Yiu WH. Innate immunity in diabetic kidney disease. Nat Rev Nephrol. 2020;16(4):206–222. doi:10.1038/s41581-019-0234-4
32. Fortpied J, Vertommen D, Van Schaftingen E. Binding of mannose-binding lectin to fructosamines: a potential link between hyperglycaemia and complement activation in diabetes. Diabetes Metab Res Rev. 2010;26(4):254–260. doi:10.1002/dmrr.1079
33. Flyvbjerg A. The role of the complement system in diabetic nephropathy. Nat Rev Nephrol. 2017;13(5):311–318. doi:10.1038/nrneph.2017.31
34. Moon JY, Jeong KH, Lee TW, Ihm CG, Lim SJ, Lee SH. Aberrant recruitment and activation of t cells in diabetic nephropathy. Am J Nephrol. 2012;35(2):164–174. doi:10.1159/000334928
35. Ryba-Stanislawowska M, Skrzypkowska M, Mysliwiec M, Mysliwska J. Loss of the balance between cd4(+)foxp3(+) regulatory t cells and cd4(+)il17a(+) th17 cells in patients with type 1 diabetes. Hum Immunol. 2013;74:701–707. doi:10.1016/j.humimm.2013.01.024
36. Klessens CQF, Zandbergen M, Wolterbeek R, et al. Macrophages in diabetic nephropathy in patients with type 2 diabetes. Nephrol Dial Transplant. 2017;32:1322–1329. doi:10.1093/ndt/gfw260
37. Tang PM, Nikolic-Paterson DJ, Lan HY. Macrophages: versatile players in renal inflammation and fibrosis. Nat Rev Nephrol. 2019;15:144–158. doi:10.1038/s41581-019-0110-2
38. Li HD, You YK, Shao BY, et al. Roles and crosstalks of macrophages in diabetic nephropathy. Front Immunol. 2022;13:1015142. doi:10.3389/fimmu.2022.1015142
39. Lv LL, Tang PM, Li CJ, et al. The pattern recognition receptor, mincle, is essential for maintaining the m1 macrophage phenotype in acute renal inflammation. Kidney Int. 2017;91:587–602. doi:10.1016/j.kint.2016.10.020
40. Meng XM, Tang PM, Li J, Lan HY. Macrophage phenotype in kidney injury and repair. Kidney Dis. 2015;1:138–146. doi:10.1159/000431214
41. Zhang X, Yang Y, Zhao Y. Macrophage phenotype and its relationship with renal function in human diabetic nephropathy. PLoS One. 2019;14(9):e0221991. doi:10.1371/journal.pone.0221991
42. Ji L, Chen Y, Wang H, et al. Overexpression of sirt6 promotes m2 macrophage transformation, alleviating renal injury in diabetic nephropathy. Int J Oncol. 2019;55(1):103–115. doi:10.3892/ijo.2019.4800
43. Williams BM, Cliff CL, Lee K, Squires PE, Hills CE. The role of the nlrp3 inflammasome in mediating glomerular and tubular injury in diabetic nephropathy. Front Physiol. 2022;13:907504. doi:10.3389/fphys.2022.907504
44. Hou Q, Kan S, Wang Z, et al. Inhibition of hdac6 with cay10603 ameliorates diabetic kidney disease by suppressing nlrp3 inflammasome. Front Pharmacol. 2022;13:938391. doi:10.3389/fphar.2022.938391
45. Zhao J, Chen J, Li YY, Xia LL, Wu YG. Bruton’s tyrosine kinase regulates macrophage‑induced inflammation in the diabetic kidney via nlrp3 inflammasome activation. Int J Mol Med. 2021;47(4):48. doi:10.3892/ijmm.2021.4881
46. Fan Z, Wang Y, Xu X, Wu Y. Inhibitor of bruton’s tyrosine kinases, PCI-32765, decreases pro-inflammatory mediators’ production in high glucose-induced macrophages. Int Immunopharmacol. 2018;58:145–153. doi:10.1016/j.intimp.2018.03.019
47. Choi SY, Lim SW, Salimi S, et al. Tonicity-responsive enhancer-binding protein mediates hyperglycemia-induced inflammation and vascular and renal injury. J Am Soc Nephrol. 2018;29(2):492–504. doi:10.1681/ASN.2017070718
48. Bus P, Pierneef L, Bor R, et al. Apolipoprotein c-i plays a role in the pathogenesis of glomerulosclerosis. J Pathol. 2017;241(5):589–599. doi:10.1002/path.4859
49. Nitta T, Kanoh H, Inamori KI, Suzuki A, Takahashi T, Inokuchi JI. Globo-series glycosphingolipids enhance toll-like receptor 4-mediated inflammation and play a pathophysiological role in diabetic nephropathy. Glycobiology. 2019;29:260–268. doi:10.1093/glycob/cwy105
50. Ma Y, Cai F, Huang X, et al. Mannose-binding lectin activates the nuclear factor-kappab and renal inflammation in the progression of diabetic nephropathy. FASEB J. 2022;36:e22227. doi:10.1096/fj.202101852R
51. Menzies RI, Booth JWR, Mullins JJ, et al. Hyperglycemia-induced renal p2x7 receptor activation enhances diabetes-related injury. EBioMedicine. 2017;19:73–83. doi:10.1016/j.ebiom.2017.04.011
52. Goldberg R, Rubinstein AM, Gil N, et al. Role of heparanase-driven inflammatory cascade in pathogenesis of diabetic nephropathy. Diabetes. 2014;63(12):4302–4313. doi:10.2337/db14-0001
53. Yu S, Cheng Y, Li B, et al. M1 macrophages accelerate renal glomerular endothelial cell senescence through reactive oxygen species accumulation in streptozotocin-induced diabetic mice. Int Immunopharmacol. 2020;81:106294. doi:10.1016/j.intimp.2020.106294
54. Nishad R, Mukhi D, Kethavath S, et al. Podocyte derived tnf-alpha mediates monocyte differentiation and contributes to glomerular injury. FASEB J. 2022;36:e22622.
55. Haruhara K, Suzuki T, Wakui H, et al. Deficiency of the kidney tubular angiotensin ii type1 receptor-associated protein atrap exacerbates streptozotocin-induced diabetic glomerular injury via reducing protective macrophage polarization. Kidney Int. 2022;101(5):912–928. doi:10.1016/j.kint.2022.01.031
56. Yang H, Xie T, Li D, et al. Tim-3 aggravates podocyte injury in diabetic nephropathy by promoting macrophage activation via the nf-kappab/tnf-alpha pathway. Mol Metab. 2019;23:24–36. doi:10.1016/j.molmet.2019.02.007
57. Ma Y, Chen Y, Xu H, Du N. The influence of angiopoietin-like protein 3 on macrophages polarization and its effect on the podocyte emt in diabetic nephropathy. Front Immunol. 2023;14:1228399. doi:10.3389/fimmu.2023.1228399
58. Lv LL, Feng Y, Wu M, et al. Exosomal miRNA-19b-3p of tubular epithelial cells promotes m1 macrophage activation in kidney injury. Cell Death Differ. 2020;27:210–226. doi:10.1038/s41418-019-0349-y
59. Ding X, Jing N, Shen A, et al. Mir-21-5p in macrophage-derived extracellular vesicles affects podocyte pyroptosis in diabetic nephropathy by regulating a20. J Endocrinol Invest. 2021;44(6):1175–1184. doi:10.1007/s40618-020-01401-7
60. Jia Y, Chen J, Zheng Z, et al. Tubular epithelial cell-derived extracellular vesicles induce macrophage glycolysis by stabilizing hif-1alpha in diabetic kidney disease. Mol Med. 2022;28(1):95. doi:10.1186/s10020-022-00525-1
61. Zhao J, Chen J, Zhu W, Qi XM, Wu YG. Exosomal miR −7002-5p derived from high glucose-induced macrophages suppresses autophagy in tubular epithelial cells by targeting Atg9b. FASEB J. 2022;36(9):e22501. doi:10.1096/fj.202200550RR
62. Liu Y, Li X, Zhao M, et al. Macrophage-derived exosomes promote activation of nlrp3 inflammasome and autophagy deficiency of mesangial cells in diabetic nephropathy. Life Sci. 2023;330:121991. doi:10.1016/j.lfs.2023.121991
63. Liu JL, Zhang L, Huang Y, et al. Epsin1-mediated exosomal sorting of dll4 modulates the tubular-macrophage crosstalk in diabetic nephropathy. Mol Ther. 2023;31(5):1451–1467. doi:10.1016/j.ymthe.2023.03.027
64. Wang Z, Sun W, Li R, Liu Y. miRNA-93-5p in exosomes derived from m2 macrophages improves lipopolysaccharide-induced podocyte apoptosis by targeting toll-like receptor 4. Bioengineered. 2022;13(3):7683–7696. doi:10.1080/21655979.2021.2023794
65. Huang H, Liu H, Tang J, et al. M2 macrophage-derived exosomal miR −25-3p improves high glucose-induced podocytes injury through activation autophagy via inhibiting DUSP1 expression. IUBMB Life. 2020;72(12):2651–2662. doi:10.1002/iub.2393
66. Kaminski H, Couzi L, Eberl M. Unconventional t cells and kidney disease. Nat Rev Nephrol. 2021;17(12):795–813. doi:10.1038/s41581-021-00466-8
67. Casey KA, Fraser KA, Schenkel JM, et al. Antigen-independent differentiation and maintenance of effector-like resident memory t cells in tissues. J Immunol. 2012;188(10):4866–4875. doi:10.4049/jimmunol.1200402
68. Forbes JM, McCarthy DA, Kassianos AJ, et al. T-cell expression and release of kidney injury molecule-1 in response to glucose variations initiates kidney injury in early diabetes. Diabetes. 2021;70(8):1754–1766. doi:10.2337/db20-1081
69. Zhang N, Tai J, Qu Z, et al. Increased CD4 + CXCR5 + T follicular helper cells in diabetic nephropathy. Autoimmunity. 2016;49(6):405–413. doi:10.1080/08916934.2016.1196677
70. Lei L, Cui L, Mao Y, et al. Augmented cd25 and cd69 expression on circulating cd8+ t cells in type 2 diabetes mellitus with albuminuria. Diabetes Metab. 2017;43(4):382–384. doi:10.1016/j.diabet.2016.10.002
71. Lampropoulou IT, Stangou M, Sarafidis P, et al. Tnf-alpha pathway and t-cell immunity are activated early during the development of diabetic nephropathy in type ii diabetes mellitus. Clin Immunol. 2020;215:108423. doi:10.1016/j.clim.2020.108423
72. Feng Y, Zhong X, Ni HF, et al. Urinary small extracellular vesicles derived CCL21 mRNA as biomarker linked with pathogenesis for diabetic nephropathy. J Transl Med. 2021;19:355. doi:10.1186/s12967-021-03030-x
73. Jin J, Wang L, Liu Y, et al. Depiction of immune heterogeneity of peripheral blood from patients with type ii diabetic nephropathy based on mass cytometry. Front Endocrinol. 2022;13:1018608. doi:10.3389/fendo.2022.1018608
74. Meng XM, Nikolic-Paterson DJ, Lan HY. Inflammatory processes in renal fibrosis. Nat Rev Nephrol. 2014;10(9):493–503. doi:10.1038/nrneph.2014.114
75. Gharaie Fathabad S, Kurzhagen JT, Sadasivam M, et al. T lymphocytes in acute kidney injury and repair. Semin Nephrol. 2020;40(2):114–125. doi:10.1016/j.semnephrol.2020.01.003
76. Liu Y, Lv Y, Zhang T, et al. T cells and their products in diabetic kidney disease. Front Immunol. 2023;14:1084448. doi:10.3389/fimmu.2023.1084448
77. Nastase MV, Zeng-Brouwers J, Beckmann J, et al. Biglycan, a novel trigger of th1 and th17 cell recruitment into the kidney. Matrix Biol. 2018;68-69:293–317. doi:10.1016/j.matbio.2017.12.002
78. Yu H, Cui S, Mei Y, et al. Mesangial cells exhibit features of antigen-presenting cells and activate cd4+ t cell responses. J Immunol Res. 2019;2019:2121849. doi:10.1155/2019/2121849
79. Li L, Tang W, Zhang Y, et al. Targeting tissue-resident memory cd8(+) t cells in the kidney is a potential therapeutic strategy to ameliorate podocyte injury and glomerulosclerosis. Mol Ther. 2022;30(8):2746–2759. doi:10.1016/j.ymthe.2022.04.024
80. Kuo HL, Huang CC, Lin TY, Lin CY. Il-17 and cd40 ligand synergistically stimulate the chronicity of diabetic nephropathy. Nephrol Dial Transplant. 2018;33(2):248–256. doi:10.1093/ndt/gfw397
81. Balakumar P, Reddy J, Singh M. Do resident renal mast cells play a role in the pathogenesis of diabetic nephropathy? Mol Cell Biochem. 2009;330(1–2):187–192. doi:10.1007/s11010-009-0132-3
82. Zheng JM, Yao GH, Cheng Z, Wang R, Liu ZH. Pathogenic role of mast cells in the development of diabetic nephropathy: a study of patients at different stages of the disease. Diabetologia. 2012;55(3):801–811. doi:10.1007/s00125-011-2391-2
83. de Morais RB, Do Couto Muniz VP, Nunes Costa E, et al. Mast cell population in the development of diabetic nephropathy: effects of renin angiotensin system inhibition. Biomed Pharmacother. 2018;107:1115–1118. doi:10.1016/j.biopha.2018.08.066
84. Yin DD, Luo JH, Zhao ZY, Liao YJ, Li Y. Tranilast prevents renal interstitial fibrosis by blocking mast cell infiltration in a rat model of diabetic kidney disease. Mol Med Rep. 2018;17(5):7356–7364. doi:10.3892/mmr.2018.8776
85. Fan Z, Gao Y, Jiang N, Zhang F, Liu S, Li Q. Immune-related serpina3 as a biomarker involved in diabetic nephropathy renal tubular injury. Front Immunol. 2022;13:979995. doi:10.3389/fimmu.2022.979995
86. LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112(5):1570–1580. doi:10.1182/blood-2008-02-078071
87. Nicoloff G, Blazhev A, Petrova C, Christova P. Circulating immune complexes among diabetic children. Clin Dev Immunol. 2004;11(1):61–66. doi:10.1080/10446670410001670517
88. Xiao X, Ma B, Dong B, et al. Cellular and humoral immune responses in the early stages of diabetic nephropathy in nod mice. J Autoimmun. 2009;32(2):85–93. doi:10.1016/j.jaut.2008.12.003
89. Abdelsamie SA, Li Y, Huang Y, et al. Oxidized LDL immune complexes stimulate collagen iv production in mesangial cells via fc gamma receptors I and III. Clin Immunol. 2011;139(3):258–266. doi:10.1016/j.clim.2011.01.016
90. Smith MJ, Simmons KM, Cambier JC. B cells in type 1 diabetes mellitus and diabetic kidney disease. Nat Rev Nephrol. 2017;13(11):712–720. doi:10.1038/nrneph.2017.138
91. Li T, Yu Z, Qu Z, Zhang N, Crew R, Jiang Y. Decreased number of cd19(+)cd24(hi)cd38(hi) regulatory b cells in diabetic nephropathy. Mol Immunol. 2019;112:233–239. doi:10.1016/j.molimm.2019.05.014
92. Huang W, Huang J, Liu Q, et al. Neutrophil-lymphocyte ratio is a reliable predictive marker for early-stage diabetic nephropathy. Clin Endocrinol (Oxf). 2015;82:229–233. doi:10.1111/cen.12576
93. Ciray H, Aksoy AH, Ulu N, Cizmecioglu A, Gaipov A, Solak Y. Nephropathy, but not angiographically proven retinopathy, is associated with neutrophil to lymphocyte ratio in patients with type 2 diabetes. Exp Clin Endocrinol Diabetes. 2015;123(05):267–271. doi:10.1055/s-0035-1547257
94. Wang Y, Peng X, Hu J, et al. Low-dose colchicine in type 2 diabetes with microalbuminuria: a double-blind randomized clinical trial. J Diabetes. 2021;13(10):827–836. doi:10.1111/1753-0407.13174
95. Wang Y, Li M, Stadler S, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184(2):205–213. doi:10.1083/jcb.200806072
96. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191(3):677–691. doi:10.1083/jcb.201006052
97. Zheng F, Ma L, Li X, et al. Neutrophil extracellular traps induce glomerular endothelial cell dysfunction and pyroptosis in diabetic kidney disease. Diabetes. 2022;71(12):2739–2750. doi:10.2337/db22-0153
98. Gupta A, Singh K, Fatima S, et al. Neutrophil extracellular traps promote nlrp3 inflammasome activation and glomerular endothelial dysfunction in diabetic kidney disease. Nutrients;2022. 14. doi:10.3390/nu15010014
99. Kunder M, Lakshmaiah V, Moideen Kutty AV. Selective decrease in alpha1-antitrypsin levels in diabetic retinopathy: could the levels of it be playing a role in the pathophysiology of diabetic retinopathy? Indian J Med Res. 2022;156:104–110.
100. Schuijs MJ, Halim TYF. Group 2 innate lymphocytes at the interface between innate and adaptive immunity. Ann N Y Acad Sci. 2018;1417(1):87–103. doi:10.1111/nyas.13604
101. Zhu J. T helper 2 (th2) cell differentiation, type 2 innate lymphoid cell (ilc2) development and regulation of interleukin-4 (il-4) and il-13 production. Cytokine. 2015;75(1):14–24. doi:10.1016/j.cyto.2015.05.010
102. Doherty TA, Broide DH. Group 2 innate lymphoid cells: new players in human allergic diseases. J Investig Allergol Clin Immunol. 2015;25:1–11.
103. Mikami Y, Takada Y, Hagihara Y, Kanai T. Innate lymphoid cells in organ fibrosis. Cytokine Growth Factor Rev. 2018;42:27–36. doi:10.1016/j.cytogfr.2018.07.002
104. Lu P, Ji X, Wan J, Xu H. Activity of group 2 innate lymphoid cells is associated with chronic inflammation and dysregulated metabolic homoeostasis in type 2 diabetic nephropathy. Scand J Immunol. 2018;87(2):99–107. doi:10.1111/sji.12637
105. Liu C, Qin L, Ding J, et al. Group 2 innate lymphoid cells participate in renal fibrosis in diabetic kidney disease partly via TGF-β1 signal pathway. J Diabetes Res. 2019;2019:8512028. doi:10.1155/2019/8512028
106. Potter SS. Single-cell RNA sequencing for the study of development, physiology and disease. Nat Rev Nephrol. 2018;14(8):479–492. doi:10.1038/s41581-018-0021-7
107. Chen J, Luo P, Wang C, et al.Integrated single-cell transcriptomics and proteomics reveal cellular-specific responses and microenvironment remodeling in aristolochic acid nephropathy. JCI Insight. 2022;(16):7. doi:10.1172/jci.insight.157360
108. Wu H, Kirita Y, Donnelly EL, Humphreys BD. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. J Am Soc Nephrol. 2019;30(1):23–32. doi:10.1681/ASN.2018090912
109. Wilson PC, Wu H, Kirita Y, et al. The single-cell transcriptomic landscape of early human diabetic nephropathy. Proc Natl Acad Sci U S A. 2019;116(39):19619–19625. doi:10.1073/pnas.1908706116
110. Fu J, Akat KM, Sun Z, et al. Single-cell RNA profiling of glomerular cells shows dynamic changes in experimental diabetic kidney disease. J Am Soc Nephrol. 2019;30(4):533–545. doi:10.1681/ASN.2018090896
111. Fu J, Sun Z, Wang X, et al. The single-cell landscape of kidney immune cells reveals transcriptional heterogeneity in early diabetic kidney disease. Kidney Int. 2022;102(6):1291–1304. doi:10.1016/j.kint.2022.08.026
112. Wu C, Tao Y, Li N, et al. Prediction of cellular targets in diabetic kidney diseases with single-cell transcriptomic analysis of db/db mouse kidneys. J Cell Commun Signal. 2023;17(1):169–188. doi:10.1007/s12079-022-00685-z
113. Wu H, Humphreys BD. Immune cell heterogeneity in a mouse model of diabetic kidney disease. Kidney Int. 2022;102(6):1215–1216. doi:10.1016/j.kint.2022.09.007
114. Gottesfeld JM, Carey MF. Introduction to the thematic minireview series: chromatin and transcription. J Biol Chem. 2018;293(36):13775–13777. doi:10.1074/jbc.TM118.004544
115. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213–1218. doi:10.1038/nmeth.2688
116. Kuppe C, Perales-Paton J, Saez-Rodriguez J, Kramann R. Experimental and computational technologies to dissect the kidney at the single-cell level. Nephrol Dial Transplant. 2022;37(4):628–637. doi:10.1093/ndt/gfaa233
117. Stoeckius M, Hafemeister C, Stephenson W, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14(9):865–868. doi:10.1038/nmeth.4380
118. Yao X, Shen H, Cao F, et al. Bioinformatics analysis reveals crosstalk among platelets, immune cells, and the glomerulus that may play an important role in the development of diabetic nephropathy. Front Med. 2021;8:657918. doi:10.3389/fmed.2021.657918
119. Zhou W, Liu Y, Hu Q, Zhou J, Lin H. The landscape of immune cell infiltration in the glomerulus of diabetic nephropathy: evidence based on bioinformatics. BMC Nephrol. 2022;23(1):303. doi:10.1186/s12882-022-02906-4
120. Ming J, Sana S, Deng X. Identification of copper-related biomarkers and potential molecule mechanism in diabetic nephropathy. Front Endocrinol. 2022;13:978601. doi:10.3389/fendo.2022.978601
121. Fu S, Cheng Y, Wang X, et al. Identification of diagnostic gene biomarkers and immune infiltration in patients with diabetic kidney disease using machine learning strategies and bioinformatic analysis. Front Med. 2022;9:918657. doi:10.3389/fmed.2022.918657
122. Jia Y, Xu H, Yu Q, Tan L, Xiong Z. Identification and verification of vascular cell adhesion protein 1 as an immune-related hub gene associated with the tubulointerstitial injury in diabetic kidney disease. Bioengineered. 2021;12(1):6655–6673. doi:10.1080/21655979.2021.1976540
123. Ye Z, Zhang Y, Huang N, Chen S, Wu X, Li L. Immune repertoire and evolutionary trajectory analysis in the development of diabetic nephropathy. Front Immunol. 2022;13:1006137. doi:10.3389/fimmu.2022.1006137
124. Li T, Shen K, Li J, Leung SWS, Zhu T, Shi Y. Glomerular endothelial cells are the coordinator in the development of diabetic nephropathy. Front Med. 2021;8:655639. doi:10.3389/fmed.2021.655639
125. Wei Y, Gao X, Li A, Liang M, Jiang Z. Single-nucleus transcriptomic analysis reveals important cell cross-talk in diabetic kidney disease. Front Med. 2021;8:657956. doi:10.3389/fmed.2021.657956
126. Mkm M, Yung S, Chan TM. Mtor inhibition and kidney diseases. Transplantation. 2018;102:S32–S40. doi:10.1097/TP.0000000000001729
127. Lu X, Li L, Suo L, et al. Single-cell RNA sequencing profiles identify important pathophysiologic factors in the progression of diabetic nephropathy. Front Cell Dev Biol. 2022;10:798316. doi:10.3389/fcell.2022.798316
128. Sur S, Nguyen M, Boada P, Sigdel TK, Sollinger H, Sarwal MM. Fcer1: a novel molecule implicated in the progression of human diabetic kidney disease. Front Immunol. 2021;12:769972. doi:10.3389/fimmu.2021.769972
129. Yu K, Li D, Xu F, et al. Ido1 as a new immune biomarker for diabetic nephropathy and its correlation with immune cell infiltration. Int Immunopharmacol. 2021;94:107446. doi:10.1016/j.intimp.2021.107446
130. Xu Q, Li B, Wang Y, et al. Identification of vcan as hub gene for diabetic kidney disease immune injury using integrated bioinformatics analysis. Front Physiol. 2021;12:651690. doi:10.3389/fphys.2021.651690
131. Chen H, Zhang Z, Zhou L, et al. Identification of ccl19 as a novel immune-related biomarker in diabetic nephropathy. Front Genet. 2022;13:830437. doi:10.3389/fgene.2022.830437
132. Li X, Liu J, Zeng M, et al. Gbp2 promotes m1 macrophage polarization by activating the notch1 signaling pathway in diabetic nephropathy. Front Immunol. 2023;14:1127612. doi:10.3389/fimmu.2023.1127612
133. Zhou M, Lu F, Jiang L, et al. Decoding the intercellular cross-talking between immune cells and renal innate cells in diabetic kidney disease by bioinformatics. J Inflamm Res. 2023;16:3049–3062. doi:10.2147/JIR.S409017
134. Li C, Su F, Zhang L, et al. Identifying potential diagnostic genes for diabetic nephropathy based on hypoxia and immune status. J Inflamm Res. 2021;14:6871–6891. doi:10.2147/JIR.S341032
135. Lu K, Wang L, Fu Y, Li G, Zhang X, Cao M. Bioinformatics analysis identifies immune-related gene signatures and subtypes in diabetic nephropathy. Front Endocrinol. 2022;13:1048139. doi:10.3389/fendo.2022.1048139
136. Zhang X, Chao P, Zhang L, et al. Single-cell RNA and transcriptome sequencing profiles identify immune-associated key genes in the development of diabetic kidney disease. Front Immunol. 2023;14:1030198. doi:10.3389/fimmu.2023.1030198
137. Xu M, Zhou H, Hu P, et al. Identification and validation of immune and oxidative stress-related diagnostic markers for diabetic nephropathy by wgcna and machine learning. Front Immunol. 2023;14:1084531. doi:10.3389/fimmu.2023.1084531
138. Chen J, Luo SF, Yuan X, et al. Diabetic kidney disease-predisposing proinflammatory and profibrotic genes identified by weighted gene co-expression network analysis (wgcna). J Cell Biochem. 2022;123(2):481–492. doi:10.1002/jcb.30195
139. Ma J, Li C, Liu T, et al. Identification of markers for diagnosis and treatment of diabetic kidney disease based on the ferroptosis and immune. Oxid Med Cell Longev. 2022;2022:9957172. doi:10.1155/2022/9957172
140. Wang Y, Zhao M, Zhang Y. Identification of fibronectin 1 (fn1) and complement component 3 (c3) as immune infiltration-related biomarkers for diabetic nephropathy using integrated bioinformatic analysis. Bioengineered. 2021;12(1):5386–5401. doi:10.1080/21655979.2021.1960766
141. Han H, Chen Y, Yang H, et al. Identification and verification of diagnostic biomarkers for glomerular injury in diabetic nephropathy based on machine learning algorithms. Front Endocrinol. 2022;13:876960. doi:10.3389/fendo.2022.876960
142. Zhang H, Hu J, Zhu J, Li Q, Fang L. Machine learning-based metabolism-related genes signature and immune infiltration landscape in diabetic nephropathy. Front Endocrinol. 2022;13:1026938. doi:10.3389/fendo.2022.1026938
143. Zhou H, Mu L, Yang Z, Shi Y. Identification of a novel immune landscape signature as effective diagnostic markers related to immune cell infiltration in diabetic nephropathy. Front Immunol. 2023;14:1113212. doi:10.3389/fimmu.2023.1113212
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.