")
Back to Journals » International Journal of Nanomedicine » Volume 19
An Immunoreceptor-Targeting Strategy with Minimalistic C3b Peptide Fusion Enhances SARS-CoV-2 RBD mRNA Vaccine Immunogenicity
Authors Chiu CT, Tsai HH, Chen JY, Hu CMJ , Chen HW
Received 15 February 2024
Accepted for publication 11 July 2024
Published 17 July 2024 Volume 2024:19 Pages 7201—7214
DOI https://doi.org/10.2147/IJN.S463546
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yan Shen
Chun-Ta Chiu,1 Hsiao-Han Tsai,2 Jing-Yuan Chen,1,3 Che-Ming Jack Hu,2 Hui-Wen Chen1,3
1Department of Veterinary Medicine, National Taiwan University, Taipei, 10617, Taiwan; 2Institute of Biomedical Sciences, Academia Sinica, Taipei, 115, Taiwan; 3Animal Resource Center, National Taiwan University, Taipei, 10617, Taiwan
Correspondence: Hui-Wen Chen, Department of Veterinary Medicine, National Taiwan University, 1 Sec 4 Roosevelt Road, Taipei, 10617, Taiwan, Tel +886 2 33669450, Email [email protected]
Introduction: The clinical success of mRNA vaccine during the COVID-19 pandemic has inspired emerging approaches to elevate mRNA vaccine immunogenicity. Among them, antigen fusion protein designs for improved immune cell targeting have been shown to augment humoral immunity against small antigen targets.
Methods: This research demonstrates that SARS-CoV-2 receptor binding domain (RBD) fusion with a minimalistic peptide segment of complement component 3b (C3b, residues 727– 767) ligand can improve mRNA vaccine immunogenicity through antigen targeting to complement receptor 1 (CR1). We affirm vaccines’ antigenicity and targeting ability towards specific receptors through Western blot and immunofluorescence assay. Furthermore, mice immunization studies help the investigation of the antibody responses.
Results: Using SARS-CoV-2 Omicron RBD antigen, we compare mRNA vaccine formulations expressing RBD fusion protein with mouse C3b peptide (RBD-mC3), RBD fusion protein with mouse Fc (RBD-Fc), and wild-type RBD. Our results confirm the proper antigenicity and normal functionality of RBD-mC3. Upon validating comparable antigen expression by the different vaccine formulations, receptor-targeting capability of the fusion antigens is further confirmed. In mouse immunization studies, we show that while both RBD-mC3 and RBD-Fc elevate vaccine immunogenicity, RBD-mC3 leads to more sustained RBD-specific titers over the RBD-Fc design, presumably due to reduced antigenic diversion by the minimalistic targeting ligand.
Conclusion: The study demonstrates a novel C3b-based antigen design strategy for immune cell targeting and mRNA vaccine enhancement.
Keywords: vaccinology, nanotechnology, complement component 3b, follicular dendritic cells, complement receptor 1, immune cell targeting
Graphical Abstract:
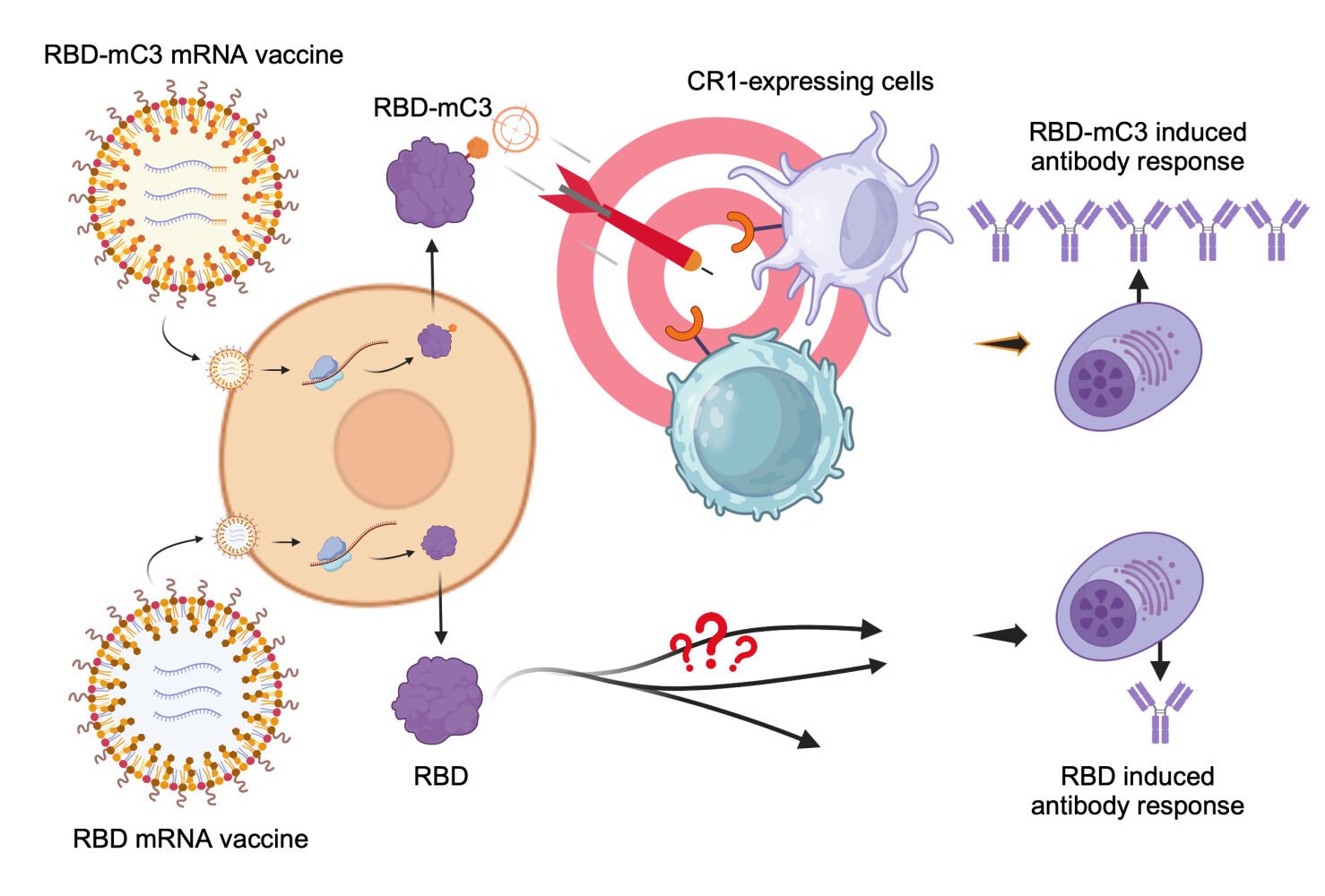
Introduction
Global efforts to contain the COVID-19 pandemic has brought forth development of novel diagnostic methods and vaccine strategies.1–3 In particular, clinical approval of the first mRNA vaccine, BNT162b from Pfizer/BioNTech, highlights numerous advantages of the nucleic-acid-based formulations against pressing infectious threats.4 In addition to bearing inherent immunogenicity conducive to adaptive immunity induction, the rapid synthesis and cost-effective production of lipid nanoparticle-based mRNA vaccines make them desirable for emergent public health responses.4–8 Their cell-free manufacturing minimizes the risk of toxin contamination. Furthermore, the mRNA is unable to integrate into the host genome, avoiding the risk of carcinogenesis and insertional mutagenesis.9 Growing enthusiasm towards this emerging vaccine technology has brought forth continuing developmental efforts to enhance mRNA vaccine immunogenicity.10,11 Among these efforts, fusion protein designs for antigen immune cell targeting have been broadly adopted to improve mRNA vaccine immunogenicity, particularly for diminutive antigen targets.11,12
Targeting antigens to immune cells strengthens humoral immune responses by enhancing antigen delivery and presentation. For instance, targeting receptors on the surface of immune cells, such as Fc receptors (FcR), CD40, and DEC-205 (CD205), has been shown to strongly enhance immune responses in vaccine development against influenza virus, human immunodeficiency virus (HIV), human papillomavirus, respiratory syncytial virus (RSV), and SARS-CoV-2.13–17 Despite a large body of literature reporting the benefits of immune cell targeting fusion protein design for vaccine development, few examples have entered clinical trials as safety concerns linger. The use of large immune cell targeting ligands derived from endogenous proteins as vaccine components raises concerns of autoimmunity.18 In addition, these ligands may also lead to antigenic diversion that distracts humoral responses from neutralizing epitopes.19 In light of these considerations, we herein explore a minimalistic 41-mer C3b peptide (residues 727–767) as a novel antigen fusion protein design strategy for immune cell targeting. We employ the design on a mRNA vaccine targeting SARS-CoV-2 receptor binding domain (RBD) with the aim of directing the small RBD antigens to complement receptor 1 (CR1) for immunogenicity enhancement.
CR1, also known as CD35, is mainly expressed on the surface of B cells and follicular dendritic cells (FDCs).20 Interaction between CR1 and complement components allows for prominent uptakes of circulating immune complexes and the subsequent regulation of humoral immunity.21,22 It mediates the modulation of inflammatory responses and the selection and maturation of B cells.23 The lack of CR1 expression has been shown to cause a significant reduction in germinal center sizes and numbers.24 Moreover, FDCs possess an essential characteristic of antigen retention, promoting B cell somatic hypermutation, antibody affinity maturation, and long-term memory of humoral immunity.25,26 We propose that vaccines directed at CR1 can elicit long-lasting humoral responses. Previous research indicates that CR1 interacts with C3b specifically within amino acid residues 727–767,27–30 suggesting the potential of this tiny C3b peptide as a precise tool for aiming at the CR1 accurately. Consequently, we combine the SARS-CoV-2 Omicron spike RBD with the C3b peptide (residues 727–767) to create an innovative mRNA vaccine platform that targets immune cells. This C3b-based tactic is herein compared to vaccine formulations with non-modified RBD and commonly adopted Fc fusion antigen design. Antigen expression, receptor targeting, and long-term humoral responses are examined for the different mRNA vaccine formulations.
Materials and Methods
Cell Culture
The human embryonic kidney (HEK) 293T cell line was maintained at 37°C and 5% CO2 in Dulbecco’s modified Eagle medium (DMEM) (Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (Gibco) and 1% Antibiotic Antimycotic Solution (Sigma-Aldrich, St. Louis, MO), and was subcultured twice a week. HEK293T cell line was purchased commercially from Riken Bioresource Center (Tsukuba, Ibaraki, Japan).
Construction of Plasmids
The targeting ligands, mC3 (GenBank accession #NP_033908.2| residues 727–767) and mFc (GenBank accession #ASY03226.1| residues 240–461), were selected and fused to the carboxyl terminus of SARS-CoV-2 Omicron (B.1.1.529) spike RBD (GenBank accession #YP_009724390.1| residues 322–544) with a glycine linker (GGGGS). The signal peptide, MFVFLVLLPLVSSQCV (GenBank accession #QRW65468.1), was derived from the spike glycoprotein.1 Genes that encoded designed antigens were synthesized by Biotools (New Taipei, Taiwan) and codon-optimized for human (Homo sapiens). The plasmids used for mRNA production were synthesized by RNA Technology Platform and Gene Manipulation Core at Academia Sinica in Taiwan. Plasmid concentrations were measured by NanoDrop Spectrophotometer (Thermo Scientific, Waltham, MA). Complete constructs were provided in Supplementary Figure S1.
In vitro Transcription (IVT) and mRNA Purification
The plasmids were linearized by the restriction enzyme MluI-HF (New England BioLabs, NEB, Ipswich, MA) and purified by GenepHlow Gel/PCR kit (Geneaid Biotech, New Taipei, Taiwan) before IVT. The T7 RiboMAX Large Scale Production System (Promega, Madison, WI) was mainly used for mRNA production. However, some reagents in the kit, such as the Ribo m7G Cap Analog and the uridine triphosphate (UTP), were replaced by CleanCap® Reagent AG (TriLink, San Diego, CA) and N1-methyl pseudouridine (TriLink) to enhance the capping efficiency and the mRNA stability. Furthermore, RNaseOut Recombinant Ribonuclease Inhibitor (Invitrogen, Carlsbad, CA) was added to lower the risk of mRNA damage. The working concentration of each reagent was calculated according to the manufacturers’ instructions. After overnight incubation, the mixtures were incubated with RQ1 RNase-free DNase (Promega) at 37°C for 15 min to remove the DNA templates. The mRNAs were then purified by RNeasy® Mini Kit (Qiagen, Venlo, The Netherlands), quantified by NanoDrop spectrophotometer (Thermo Scientific), and stored at −80°C.
Denaturing Gel Electrophoresis
Nuclease-free agarose powder (Bioman Scientific, New Taipei, Taiwan) was mixed and heated in RNase-free 1 × Tris-Acetate-EDTA (TAE) buffer (Invitrogen) to prepare the 1.5% agarose gel. The ssRNA ladder (NEB) and the mRNA samples were combined with RNA loading dye (NEB) (ratio 1: 4) respectively and were heated at 90°C for 2 min. Cooled down on the ice for 1 min, the denaturing RNAs were loaded on the agarose gel. Finally, the mRNAs were examined by Molecular Imager® ChemiDoc XRS+ Imaging System (Bio-Rad Laboratories, Hercules, CA) and Image Lab software (Bio-Rad Laboratories).
Lipid Nanoparticle (LNP) Formulations
Ionizable lipid [(6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl] 4-(dimethylamino) butanoate (Dlin-MC3-DMA; #34365) and 1,2-Dimyristoyl-rac-glycero-3-PE-methoxy-Polyethyleneglycol-2000 (DMPE-2kPEG; #34649) were purchased from Cayman Chemical. Cholesterol was purchased from Sigma Aldrich (St. Louis, MO). 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) was purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). LNP was produced under RNase-free conditions. First, the glass bottle and stir bar were washed with chloroform and dried with nitrogen gas. 100 µg of mRNA was added to 600 µL of 50 mM citrate buffer (Sigma-Aldrich, St. Louis, MO) in the bottle. The mRNA solution was then mixed with the 200 µL of ethanolic lipid solution and stirred simultaneously. The lipid formulation was composed of DLin-MC3-DMA, DSPC, cholesterol, and DMPE-2kPEG at a ratio of 50: 10: 38.5: 1.5.31,32 After evaporation for 4 hr in a vacuum chamber under gentle stirring, the solution was diluted in 10% sucrose buffer (Fisher Scientific, Waltham, MA) and condensed with the Amicon® centrifugal filter with a 100 kDa molecular-weight cut-off (Millipore Sigma, Burlington, MA). (Merck, Darmstadt, Germany) at 600 × g at 4°C, the mRNA-LNPs were stored at −80°C with a final concentration from 0.25 to 1.00 µg/µL and kept from repeated thawing and freezing.
mRNA Encapsulation Efficiency
The mRNA was extracted from LNP as previously reported.33 Briefly, mRNA-LNP was mixed with 60 mM ammonium acetate (Sigma) diluted in isopropanol at a 1:9 ratio. The mixture was centrifuged at 16,000× g at 4°C for 15 min, and the pellet was washed with isopropanol. The solution was then centrifuged again for 5 min. The extracted mRNA pellet was resuspended in Ambion Nuclease-Free Water (Invitrogen) and quantified by NanoDrop spectrophotometer (Thermo Scientific).
LNP Observation via Cryo-Electron Microscope (Cryo-EM)
Cryo-EM was applied for LNP observation. First, a glow-discharged grid was used to hold 10 μL of nanoparticles. The droplet was left on the grid for 10 sec before being removed with filter paper. Excess nanoparticles were washed away with distilled water loaded onto the grid. The Tecnai G2 F20 transmission electron microscope (FEI, Hillsboro, Oregon) was used with an acceleration voltage of 200 kV.
Cell Transfection
One day before transfection, 2.5×106 293T cells in 2 mL DMEM were seeded on a Corning 60 mm TC-treated Culture Dish (Fisher Scientific), or 1.0×106 293T cells/well in 1.5 mL DMEM were seeded on a Costar® 6-well plate (Fisher Scientific). The mRNA transfection was performed using Lipofectamine 3000 Transfection Reagent (Invitrogen) and was incubated at 37°C. After 48 hr, the cell lysates were harvested with 1× protein loading reducing sample buffer (Omics Biotechnology, New Taipei, Taiwan) supplemented with protease inhibitor (Sigma), followed by 5-minute intermittent sonication.
SDS-PAGE and Western Blot
Whole-cell lysates from transfected 293T cells were examined by Western blot. In brief, samples were heated at 95°C for 5 min and run on a 10% polyacrylamide gel, which was made from TGX Stain-Free FastCast Acrylamide Kit (Bio-Rad Laboratories) at 168 V for 45 min. Then, the proteins were transferred to the methanol pre-incubated Immun-Blot® Polyvinylidene Difluoride (PVDF) membrane (Bio-Rad Laboratories) at 395 mA for 1 hr. After the membrane was blocked with 5% Difco skim milk (BD, Franklin Lakes, NJ) in PBST for 30 min, proteins were detected by rabbit anti-RBD polyclonal antibody (Sino Biological #40591-T62, Beijing, China) (1:5000) in the blocking buffer for 1 hr. After washing with PBST 3 times, the membrane was incubated with HRP-conjugated goat anti-rabbit IgG (1:2000) for 1 hr at room temperature. Then, the membrane was evaluated using Clarity Western ECL Substrate (Bio-Rad Laboratories) and photographed by Molecular Imager® ChemiDoc XRS+ Imaging System (Bio-Rad Laboratories). The Omicron B.1.1.529 spike RBD recombinant protein (Sino Biological #40592-V08H121) was used as the positive control. GAPDH was recognized by GAPDH Monoclonal Antibody (6C5) (Invitrogen) (1:5000) and presented as the loading control. Experiments were performed with three replicates.
Detection of Vaccine Antigen in mRNA-LNP Transfected Cells by Immunofluorescence Assay (IFA)
One day prior to transfection, 2.0×104 293T cells/well were seeded on the Corning® CellBIND® 96-well plate (Fisher Scientific). The mRNA-LNP was transfected into cells and incubated at 37°C for 48 hr. For cell fixation, 80% acetone (Merck) was used and incubated at −20°C for 20 min. After acetone was removed, cells were blocked with 10% goat serum (Jackson ImmunoResearch, Bar Harbor, Maine) for 30 min at room temperature. Afterward, the primary antibody rabbit anti-RBD polyclonal antibody (Sino Biological #40591-T62), which was 500-fold diluted, reacted with the expressed antigens for 1 hr at room temperature. After washing with PBST three times, fluorescein (FITC) goat anti-rabbit IgG (Jackson ImmunoResearch #111-095-003) (1:2000) was incubated at room temperature for 1 hr in the dark. After washing with PBST three times, cells were stained with KPL DAPI solution (SeraCare Life Sciences, Milford, MA) (1:400) and incubated for 15 min in the dark. Finally, fluorescence was observed under the Olympus fluorescence microscope IX83 (Olympus Life Science, Tokyo, Japan), and the fluorescence integrated density was quantified using ImageJ software (version 2.14.0/1.54f) as reported previously.34 Experiments were performed with three replicates.
Binding Test of Immune Cell Receptors and Targeting Ligands
For antigen expression, transfection was performed on the Corning® CellBIND® 96-well plate (Fisher Scientific), which was seeded 2.0×104 293T cells in each well on the previous day. Transfection was conducted using Lipofectamine 3000 (Invitrogen), following the manufacturer’s instructions. The final mixture was added to each well and incubated at 37°C for 24 hr. After the antigens were expressed, the spent medium was aspirated, and the cells were fixed by 80% acetone (Merck) for 20 min at −20°C. Then, the cells were blocked with 10% goat serum (Jackson ImmunoResearch) for 1 hr at room temperature, followed by one-hour incubation of receptor proteins, including recombinant human CD35 protein (CR1) (R&D Systems #5748-CD-050, NE Minneapolis, MN) and mouse CD64/FCGR1 protein (Sino Biological #50086-M08H). Afterward, the primary antibodies, including rabbit CD35 polyclonal antibody (Invitrogen #PA5-114324) (1:500), rabbit CD64/FCGR1 monoclonal antibody (Sino Biological #50086-R027) (1:100), and mouse SARS-CoV-2 S1 monoclonal antibody (Sino Biological #40591-MM42) (1:500), were incubated for 1 hr. After washing with PBST three times, the secondary antibodies, Cy5 goat anti-mouse IgG (Jackson ImmunoResearch #115-175-146) (1:2000) and FITC goat anti-rabbit IgG (Jackson ImmunoResearch #111-095-003) (1:2000), were incubated for 1 hr and kept in dark. After washing again, the cells were stained with KPL DAPI solution (SeraCare Life Sciences) (1:400) and incubated for 15 min in the dark. The plate was washed thrice with PBST, covered by glycerol, and observed via the Olympus fluorescence microscope IX83 (Olympus Life Science). Quantification of fluorescence integrated density and colocalization analysis were investigated by ImageJ software (version 2.14.0/1.54f), and colocalization efficiency was calculated using the BIOP JACoP plugin.34,35 Experiments were performed with three replicates.
Mouse Immunization Studies
In the animal studies, 5-week-old female C57BL/6 mice were purchased from National Laboratory Animal Center and subsequently bred and housed at Animal Resource Center, National Taiwan University. Before the experiments started, mice were quarantined and acclimated to the environment for 1 week. The mice were randomly assigned to each experimental group from the start of the experiment. Seven vaccine groups were designed: RBD-mC3 (1 and 10 µg), RBD-mFc (1 and 10 µg), RBD (1 and 10 µg), and PBS group. Mice were immunized with mRNA-LNPs with 29-gauge Ultra-Fine Insulin Syringes (BD) in the right hindlimbs via the intramuscular route. The mRNA-LNPs were diluted in sterile PBS to the final volume of 50 µL for each injection. As for the vaccination schedule, the mice were primed on day 0 and boosted on day 21 and day 49. Submandibular blood collections were performed on days 21, 42, 63, and 105 post-primary immunization with the 4 mm or 5 mm point length of Goldenrod Animal Lancets (MEDIpoint, Mineola, NY). After the blood was centrifuged at 3000 × g for 10 min at 4°C, the serum was harvested and stored at −20°C. At the end of the experiment, the mice were humanely sacrificed.
Enzyme-Linked Immunosorbent Assay (ELISA)
SARS-CoV-2 Omicron RBD-specific antibody was evaluated via ELISA. Spike RBD recombinant proteins of Omicron sublineage B.1.1.529 (Sino Biological #40592-V08H121) were coated on 96-well plates (Thermo Scientific) at a concentration of 100 ng in 100 µL per well. After blocking with 5% skim milk (BD) in PBST for 1 hr at room temperature, the serially diluted serum samples were added to relative wells and incubated at room temperature for 1 hr. The serum samples were removed and the wells were washed with PBST 3 times. Then, the HRP-conjugated goat anti-mouse IgG (Abcam # ab205719) (1:2000) was added to wells and incubated at room temperature for 1 hr. After removing the secondary antibody and 3 times PBST wash, KPL SureBlue Reserve TMB Microwell Peroxidase Substrate (SeraCare Life Sciences) was incubated at room temperature for 10 min. Finally, 1 M H2SO4 stop solution was used to terminate the reaction, and the plates were recorded immediately at a wavelength of 450 nm via Synergy H1 Microplate Reader (Agilent BioTek, Winooski, VT). The optical density was read by Gen5 software (Agilent BioTek), and each sample is shown as a curve. The endpoint titers were calculated by regression analysis with a cut-off value of 0.2.
Statistical Analysis
The data were analyzed with Prism 9 (GraphPad, San Diego, CA) and Excel (Microsoft, Redmond, WA) software. The antibody titers from different vaccines were compared at different time points. Two-way ANOVA was applied to perform the statistical analysis of antibody titers. One-way ANOVA was used for the comparison of fluorescence integrated density. Data are presented as median with standard deviation (SD) and analyzed through Tukey’s multiple comparison test. Non-specific (ns) P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. When P-value was lower than 0.05, a significant difference existed.
Results
Design and Production of Immune Cell Targeting mRNA Vaccines
The spike RBD of SARS-CoV-2 plays a crucial role in the generation of neutralizing antibodies. In this study, RBD from the SARS-CoV-2 Omicron variant B.1.1.529 was used as the vaccine antigen. To improve immunogenicity, genes encoding targeting ligands, including murine C3b (mC3) and murine IgG1 Fc (mFc), were added to the 3’ end of the RBD sequence. An RBD without any ligand was also created as the control group (Figure 1A). The involvement of two kinds of ligands was aimed to make some comparisons, including their immunogenicity and targeting ability. The ligands were fused to RBD with a glycine linker (GGGGS) to ensure proper protein structures. Furthermore, a signal peptide, MFVFLVLLPLVSSQCV, from SARS-CoV-2 surface glycoprotein was employed to enhance the expression level of the vaccine antigens.1 A Kozak sequence was included to enhance ribosome recognition of the start codon.36 The 5’ UTR and 3’ UTR encoded the mouse β-globin and hemoglobin α-globin respectively. The long polyA tail, (60A)G(60A), was composed of two segments of 60 continuous adenosines and a single guanine between them. This design, similar to the A30(GCATATGACT)A70 design from BNT162b2,37,38 increased translation efficiency, enhanced structural stability, and reduced spontaneous deletion during plasmid replication.39,40 We utilized these state-of-the-art designs to maximize translation efficiency and enable strong stability. To produce proper vaccines, linearized plasmids were used as DNA templates for in vitro transcription. Synthesized RBD-mC3, RBD-mFc, and RBD mRNAs presented clear bands with sizes at 1164, 1686, and 1023 bases respectively in denatured single-stranded RNA electrophoresis (Figure 1B). Each mRNA included the components from the T7 promoter to the long polyA tail. The mRNA cannot enter the cells by itself due to its natural properties. It is also vulnerable to the extracellular environment and is easily recognized and destroyed by host immune systems.4 Therefore, mRNA was formulated into lipid nanoparticles (LNPs) for successful and efficient mRNA delivery. The mRNA encapsulation efficiency was around 98% (Supplementary Table S1). To confirm that the LNPs presented excellent quality and uniform morphology, the nanoparticles were imaged using cryo-EM. They demonstrated clear and smooth contours with an averaged diameter of approximately 100 nm (Figure 1C). Finally, three potential antigens, RBD-mC3, RBD-mFc, and RBD, were investigated in subsequent antigenicity and immunogenicity tests.
 |
Figure 1 Design and production of immune cell targeting mRNA-LNP vaccines. (A) Designs and sketches of mRNAs and immunogens: RBD, RBD-mC3, and RBD-mFc. The mRNA was composed of a Kozak sequence (Kz), a signal peptide (SP), a SARS-CoV-2 Omicron RBD with or without a targeting ligand, and a 120-adenosine segmented polyA tail. (B) The in vitro transcription of three mRNAs. The mRNAs were presented in the denaturing single-stranded RNA electrophoresis. (C) The cryo-EM image of lipid nanoparticles (LNPs). |
Validation of Vaccine Antigenicity Through HEK293T Transfection
To examine the functionality of mRNA and the antigenicity of expressed RBD antigens, HEK293T cells were transfected with each mRNA using the Lipofectamine 3000 transfection reagent for antigen expression. In addition, to evaluate the transfection of nanoparticles and the antigenicity of non-reduced antigens, we performed IFA by using anti-RBD antibodies to recognize the well-expressed antigens after mRNA-LNPs were delivered to HEK293T cells (Figure 2A). Following this, the antigens underwent Western blot analysis. RBD-mC3, RBD-mFc, and RBD were all detected by anti-RBD antibodies and exhibited distinct and strong bands with approximate molecular weights of 40 kDa, 60 kDa, and 35 kDa respectively. The RBD (without any targeting ligand) showed the same size as the purchased recombinant homologous Omicron RBD (PC). The protein sizes of RBD-mC3 and RBD-mFc surpassed that of RBD due to the fusion with targeting ligands. The approximate sizes of the two ligands, mC3 and mFc, were around 5 kDa and 25 kDa, respectively. Despite the existence of fusing ligands, the anti-RBD antibodies were still able to interact with RBD normally (Figure 2B). According to these Results, three antigens presented correct antigenicity and were well-recognized by anti-RBD antibodies under reducing conditions. Based on the IFA results, each mRNA vaccine was successfully delivered into cells and translated into antigen properly. These expressed antigens were then identified by FITC fluorescence (green), while the blue signals represented the cell nuclei. Compared to the cells without mRNA-LNP vaccine transfection, the other three panels displayed prominent antigen expressions with scattered green fluorescence across the images. Green emissions were located in the cells and appeared ostensibly separate from the nuclei (Figure 2C). Furthermore, we quantified the fluorescence integrated density of each photo with ImageJ software. Three different antigen displayed high integrated intensity ranged from 106 to 107, while no fluorescence was detected from non-transfected cell. All the mRNA-LNP vaccines presented comparable levels of fluorescence in our experiments, indicating consistent in vitro antigen expression of each vaccine (Figure 2D). These data demonstrated the feasibility of mRNA-LNP delivery. It also verified that the antigens with native forms were able to exhibit correct conformation, bind to RBD-specific antibodies, and display proper functionality.
 |
Figure 2 Validations of vaccine antigenicity via Western blot and immunofluorescence assay (IFA). (A) Schematic of antigenicity validation. The antigens were expressed via lipofectamine transfection into 293T cells and were analyzed by Western blot. The mRNA-LNP vaccines were directly transfected into 293T cells and examined by IFA. (B) In Western blot, the expressed RBD-mC3, RBD-mFc, and RBD were detected by anti-RBD polyclonal antibodies. Homologous SARS-CoV-2 Omicron RBD recombinant protein was the positive control (PC). GAPDH was the loading control. (C) In IFA, antigens were labeled by green fluorescence (FITC). Cell nuclei were stained by DAPI and presented blue fluorescence. Cells without any transfection were the negative control. (D) Expression levels of three antigens were presented as fluorescence integrated density. Data are presented as median with SD and analyzed through one-way ANOVA Tukey’s multiple comparison test. Non-specific (ns) P > 0.05. |
Potent Targeting Capability of mC3 Ligand
To understand the interaction between the receptors and their ligands, we constructed an IFA experiment to imitate the targeting process. In brief, after the mRNA-transfected HEK293T cells expressed antigens, the commercial recombinant receptors were incubated with the cells for protein targeting. The expressed RBD-mC3 and RBD-mFc were detected by anti-SARS-CoV-2 RBD antibody and stained by Cy5 fluorescence (purple). The CR1 and FcγR, which bound to the mC3 and mFc respectively, were recognized by the anti-receptor antibodies and stained by FITC fluorescence (green) (Figure 3A). During the investigation of the interaction between RBD-mC3 and CR1, the green fluorescence indicated the presence of CR1, while RBD-mC3 expression appeared as purple within the cells. Both signals depicted the contour of the cell and were uniformly distributed in the cytoplasmic region. The merged panel revealed the exclusive coexistence of CR1 with RBD-mC3 (Figure 3B). In this situation, the RBD-mC3 was initially expressed, followed by subsequent conjugation with CR1, ultimately recognized by the fluorescence markers. We performed the colocalization analysis with ImageJ software to measure the correlation between RBD-mC3 and CR1. The correlation was shown as Pearson’s coefficient, which indicated perfect colocalization when the value is 1. In this analysis, Pearson’s coefficient ranged from 0.802 to 0.856, suggesting a high degree of colocalization and effective targeting ability of RBD-mC3 to CR1. (Figure 3C). In the analysis of the interaction between RBD-mFc and FcγR, the green signals displayed the existence of FcγR, while the purple ones represented the expression of RBD-mFc. The cells were outlined by the fluorescence signals, which showcased the locations of RBD-mFc and FcγR. In the FcγR/RBD-mFc panel, two different fluorescences were detected in the same area. Upon merging with the image of nuclei (blue), we demonstrated that the FcγR (green) only existed within the cells expressing RBD-mFc (purple). The Cy5 and FITC signals showed high consistency in the distribution (Figure 3D). This result demonstrated a strong interaction between mFc and FcγR. In the colocalization analysis, Pearson’s coefficient was calculated to be 0.930 to 0.963, indicating a high degree of colocalization between FcγR and RBD-mFc (Figure 3E). From these results, we demonstrated the simulated targeting process and affirmed the targeting proficiency of RBD-mC3 and RBD-mFc towards their receptors. Here, the mC3 ligand exhibited its strong potential for immune cell targeting applications.
 |
Figure 3 Confirmation of targeting proficiency for mC3 and mFc ligands via immunofluorescence assay (IFA). (A) Illustration of the ligands-receptors interactions that were demonstrated by IFA. Following mRNA transfection into 293T cells, receptors were incubated with the cells. Then, RBD-mC3 bound CR1 while RBD-mFc bound FcγR. The RBD-mC3 and RBD-mFc were labeled by anti-RBD monoclonal antibodies. The CR1 and FcγR were marked using anti-CR1 polyclonal antibodies and anti-FcγR monoclonal antibodies, respectively. (B) Antibodies targeting CR1 (FITC, green) and RBD-mC3 (Cy5, purple) depicted the respective distributions of CR1 and RBD-mC3. (C) Colocalization analysis of RBD-mC3 and CR1. (D) Antibodies against FcγR (green) and RBD-mFc (purple) showed the distributions of FcγR and RBD-mFc respectively. The blue color (DAPI) represented the cell nuclei. (E) Colocalization analysis of RBD-mFc and FcγR. Three regions of interest (ROI) were analyzed. Pearson’s coefficient (R) was calculated in colocalization analysis. R > 0.8 suggests a very strong correlation. |
Significant Enhancement of Humoral Immune Responses with a Low Dosage in RBD-mC3 Vaccine-Administrated Mice
To validate the immune cell targeting property of mC3 to RBD antigen, we performed a three-dose vaccination in C57BL/6 mice. Humoral immune responses were compared with those induced by RBD-mFc and RBD vaccines. Mice were divided into seven groups, and the serum samples were collected on days 21, 42, 63, and 105 for antibody analysis after they were given three doses of mRNA vaccines (Figure 4A). Each mRNA vaccine was able to successfully boost the antibody responses with only two doses. In the 10 µg vaccine groups (Figure 4B), both RBD-mC3 and RBD-mFc vaccines rapidly stimulated antibody responses, with titers at 1:14 and 1:11 respectively on day 21, whereas the RBD vaccine only induced 1:4 titer. On day 42, antibody responses elicited by both immune cell targeting vaccines presented significantly higher levels (1:38 for RBD-mC3 and 1:58 for RBD-mFc), whereas the RBD vaccine displayed a lower titer at 1:21. After three doses of immunization, the RBD-mC3 vaccine elicited a titer at 1:55 at day 63 and the antibodies remain stable until day 105. We found that the RBD-mFc vaccine slightly decreased on day 105 (1:35), which was different from the trend of the RBD-mC3 vaccine (Figure 4B), suggesting durable antibodies elicited by RBD-mC3 antigen.
 |
Figure 4 Immunogenicity study of immune cell targeting mRNA vaccines in mice with low dosages. (A) Immunization schedule and vaccination groups. Mice were administrated with three doses of vaccines (1 µg or 10 µg) via the intramuscular route. Mice in the negative control group (NC) were injected with PBS. (B) Comparison of anti-RBD IgG titers from 10 µg groups. (C) Comparison of anti-RBD IgG titers from 1 µg groups. ELISA was conducted to assess the antibody levels in the sera using plates coated with recombinant homologous RBD proteins. Data are presented as median with SD and analyzed through two-way ANOVA Tukey’s multiple comparison test. Orange asterisk displayed the significance between RBD-mC3 and RBD vaccines. Blue asterisk and ns showed the analysis between RBD-mFc and RBD vaccines. Non-specific (ns) P > 0.05, *P < 0.05, **P < 0.01. |
Among 1 µg vaccination groups (Figure 4C), the RBD-mC3 vaccine induced the highest titer at 1:37 after the second vaccination. In addition, its antibody responses maintained at high levels for more than 3 months without declining. On day 105, the RBD-mC3 vaccine maintained the titers by approximately 1:50, and the titer from the RBD-mFc vaccine was at 1:43. The RBD-mC3 vaccine not only presented a superior humoral immune response among all 1 µg vaccines but also showed significance when compared to the RBD vaccine. The RBD vaccine, even with the dosage raised to 10 µg, produced low IgG titer, similar to that in 1 µg immunizations, at approximately 1:20. Conversely, the RBD-mC3 vaccine elevated antibody responses, ranging from 1:50 to 1:53. Furthermore, the titers from RBD-mC3 were about 1.5 times higher than those from RBD-mFc (1:43 at 1 µg and 1:35 at 10 µg) (Figure 4B and C). In general, the data indicated that our immune cell targeting mRNA vaccines were able to generate heightened antibody titers with only two doses of vaccination and establish enduring humoral immunity in mice. The RBD-mC3 vaccine displayed superior efficacy compared to other vaccines in both 1 µg and 10 µg immunization regimens, underscoring its remarkable targeting capability.
Discussion
The receptor-binding domain (RBD), which is located in the S1 subunit of spike protein, is the most important region for cell attachment and is the immunodominant site for the development of neutralizing antibodies against SARS-CoV-2.4,41,42 Studies have shown that a few potent and broad-effective neutralizing antibodies induced by RBD were able to conjugate all current variants.43,44 In addition, RBD barely induces antibody-dependent enhancement (ADE), which is found in full-length spike vaccines and deteriorates the infection.45,46 However, the RBD itself bears low immunogenicity and is not able to elicit enough antibody responses because of its small molecular size.47,48 Numerous research teams have dedicated efforts to developing mRNA vaccines based on the RBD.4 By employing the C3b-based fusion antigen strategy, we show significant immunogenicity improvement for RBD-based mRNA vaccine. In this study, the mC3 ligand accurately interacts with the CR1, illustrating the probable mechanism responsible for the enhanced immunogenicity. Furthermore, the mice receiving three doses of RBD-mC3 mRNA vaccines exhibited enhanced RBD-specific antibody titers surpassing those induced by the non-targeting RBD vaccine and RBD-mFc targeting vaccine. These outcomes significantly point out the successful application of the mC3 targeting ligand.
Targeting immune cells proves to be a potent and promising approach to boost the immunogenicity of vaccines. Despite numerous prior studies demonstrating the potential enhancement of vaccine-induced immunity through Fc-fused proteins,15,16,49,50 there remains uncertainty regarding whether these beneficial effects stem from the targeting capability, given the absence of direct evidence showcasing ligand-receptor interaction. It is pivotal to investigate the interaction between the targeting ligand and its relevant receptor. Moreover, as our hypothesis centers around the interaction between CR1 and mC3, it becomes essential to validate whether the mC3-based immunogen enhances humoral immunity through binding with CR1. Therefore, to address this question, we design an IFA experiment to imitate the targeting process. Our results validate several key points. First, both the mC3 and mFc ligands developed in this study are able to function normally by correctly interacting with their respective receptors. Second, the receptors solely appear within the cells harboring expressed antigens. The results of colocalization analysis show a convincing and potent correlation between fusion ligands and receptors. It suggests that the bindings between the ligands and the receptors are highly specific, devoid of any non-specific binding occurrences. Third, this experimental design provides a targeting process that is close to the real situation. Although the antigens and the receptors encounter randomly in a gigantic pool (where a small number of proteins are incubated in hundreds of microliters of medium), they engage in specific and accurate binding. All these results conclusively demonstrate the robust targeting capacity of the ligands towards the receptors.
Due to the low immunogenicity of RBD itself, mice could only reach strong immune responses via high-dose vaccinations (over 15, 30, or 42 µg) or adjuvant combinations.51,52 However, we administered low dosages (1 µg and 10 µg) without any adjuvant for mouse immunization to ascertain the efficacy of the C3b-based strategy in enhancing RBD immunogenicity. The RBD vaccine (10 µg) in our research induced a low titer at around 1:4 after the first immunization. Even following the third immunization, the titer increased only to about 1:14, which typically indicated the poor immunogenicity of RBD. In contrast, the RBD-mC3 vaccine (10 µg) elicited titers (1:55) approximately four times higher after the third immunization. Furthermore, even at a lower dosage of 1 µg, the RBD-mC3 vaccine produced a similar antibody level (titer by 1:50) after the third immunization. The RBD vaccine can barely generate helpful antibody responses. A previous study claimed that the antigens, whose sizes were below 40–50 kDa, tended to be rapidly cleared by the renal elimination.53 The limited immunogenicity of RBD (about 35 kDa) can probably be associated with its short retention in circulation. The size of RBD-mC3 (40 kDa) is also smaller than the threshold. However, after the mRNA is expressed, the RBD-mC3 antigen is secreted by the cells and targets the immune cells directly, allowing itself to bypass the swift clearance by the kidney. Hence, the C3b-based strategy can efficiently elevate the humoral responses without combining any adjuvant or increasing the dosage, highlighting its remarkable targeting ability to enhance immunogenicity.
In this study, we also construct the RBD-mFc mRNA vaccine for comparison. This experimental group serves two purposes. First, it allows us to determine the efficacy of the mFc ligand in mRNA-based vaccines. Second, it enables the simultaneous validation and comparison of various targeting strategies, thereby allowing the selection of a promising candidate. From our data, we discover a slight decrease in antibody titers induced by the RBD-mFc vaccine approximately three months after the first immunization, a phenomenon not observed in the case of the RBD-mC3 vaccine. We speculate that the RBD-mFc vaccine may stimulate lower antibody titers due to antigenic diversion, which may be a universal effect of various antigens.19,54 The RBD-specific antibodies might be exploited by the mFc ligand due to its large molecular size (25 kDa). On the contrary, the mC3 ligand with a small size (5 kDa) bears a low possibility of inducing antigenic diversion. From our data, the RBD-mC3 vaccine also displays titers approximately 1.5 times higher than those observed with the RBD-mFc vaccine after the third immunization. Therefore, the mC3 ligand, composed of only 41 amino acids, is more appropriate to be used in mRNA vaccine applications for enhancing small antigen immunogenicity with the minimum effect of antigenic diversion.
Employing the mC3 (C3b) targeting ligand for antigen fusion presents a novel design for leveraging the complement system to enhance antigen immunogenicity, contrasting with prior adjuvant research that utilized another complement component, C3d, to stimulate immunogenicity. Earlier studies developed DNA vaccines or mRNA vaccines by combining three consecutive copies of murine C3d with viral antigens, such as influenza virus hemagglutinin, human immunodeficiency virus gp120 envelope glycoprotein, measles virus hemagglutinin, and SARS-CoV-2 spike protein and RBD.11,55–57 All these vaccines effectively improved immune responses and raised antibody levels with low dosages. Compared to the repetitive C3d design, our mRNA vaccine employs a single 41-mer C3b, significantly inducing enhanced and sustained antibody responses. C3b and C3d can interact with CR1 and complement receptor 2 (CR2) respectively. Both CR1 and CR2 play pivotal roles in B cell activation, proliferation, and differentiation. They are also expressed on the surface of follicular dendritic cells (FDC), thereby prolonging the maintenance of secondary antibody responses.58 For instance, the measles virus DNA vaccine generated enduringly high levels of antibody titers, which were able to persist for over 26 weeks.56 Similarly, our RBD-mC3 mRNA vaccine can trigger long-term antibody titers for more than three months, demonstrating the robustness of the C3b fusion design. Overall, the C3b-based strategy shows potential comparable to the triple C3d design and warrants further investigation.
Conclusion
Targeting antigens to CR1 on immune cells with a minimalistic C3b peptide holds the potential for boosting the poor immunogenicity of compact antigens. In this study, the C3b-based RBD mRNA vaccine enables immunization using minimal dosages and avoids antigenic diversion concerns, significantly enhancing antibody responses compared to the unmodified RBD vaccine. Additionally, it elicits modestly higher humoral responses than the widely used Fc fusion design. Moreover, the ligand-receptor interaction experiment pioneers the investigation of the mechanism of fusion protein vaccines. This innovative vaccine C3b design provides valuable insights into the selection of targeting fusion ligands, the contribution of antigen enhancement, and the prolongation of humoral immunity.
Ethics Statement
The study adhered to relevant institutional and national regulations. The animal experiments took place at the Animal Resource Center at National Taiwan University and followed established guidelines. All mouse-related procedures were approved by the Institutional Animal Care and Use Committee (IACUC) with the approval code: No. NTU-109-EL-00067, dated 21 November 2022.
Acknowledgments
The authors thank the RNA Technology Platform and Gene Manipulation Core Facility (RNAi core) of the National Core Facility for Biopharmaceuticals at Academia Sinica in Taiwan for providing shRNA reagents and related services. Financial support from the National Science and Technology Council and National Taiwan University was greatly appreciated. The authors thank the Animal Resource Center of National Taiwan University for their dedication to animal care.
Author Contributions
All authors read and approved the final manuscript. All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
The authors thank the funding from National Science and Technology Council, Taiwan (MOST109-2327-B-002-009, MOST111-2321-B-002-017, MOST111-2124-M-001-007, NSTC112-2124-M-001-010, NSTC113-2124-M-001-018) and National Taiwan University (113L892501).
Disclosure
The authors declare no conflicts of interest in this research.
References
1. Fang E, Liu X, Li M, et al. Advances in COVID-19 mRNA vaccine development. Signal Transduct Target Ther. 2022;7(1):94. doi:10.1038/s41392-022-00950-y
2. Drobysh M, Ramanaviciene A, Viter R, et al. Biosensors for the determination of SARS-CoV-2 virus and diagnosis of COVID-19 infection. Int J Mol Sci. 2022;23(2):666.
3. Drobysh M, Ramanaviciene A, Viter R, Ramanavicius A. Affinity sensors for the diagnosis of COVID-19. Micromachines. 2021;12(4):390.
4. Chaudhary N, Weissman D, Whitehead KA. mRNA vaccines for infectious diseases: principles, delivery and clinical translation. Nat Rev Drug Discov. 2021;20(11):817–838. doi:10.1038/s41573-021-00283-5
5. Anand P, Stahel VP. Correction to: the safety of Covid-19 mRNA vaccines: a review. Patient Saf Surg. 2021;15(1):22. doi:10.1186/s13037-021-00296-4
6. Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines — a new era in vaccinology. Nat Rev Drug Discov. 2018;17(4):261–279. doi:10.1038/nrd.2017.243
7. Sahin U, Karikó K, Türeci Ö. mRNA-based therapeutics--developing a new class of drugs. Nat Rev Drug Discov. 2014;13(10):759–780. doi:10.1038/nrd4278
8. Teijaro JR, Farber DL. COVID-19 vaccines: modes of immune activation and future challenges. Nat Rev Immunol. 2021;21(4):195–197. doi:10.1038/s41577-021-00526-x
9. Le T, Sun C, Chang J, Zhang G, Yin X. mRNA vaccine development for emerging animal and zoonotic diseases. Viruses. 2022;14(2). doi:10.3390/v14020401
10. Han X, Alameh M-G, Butowska K, et al. Adjuvant lipidoid-substituted lipid nanoparticles augment the immunogenicity of SARS-CoV-2 mRNA vaccines. Nature Nanotechnol. 2023;18(9):1105–1114. doi:10.1038/s41565-023-01404-4
11. Li B, Jiang AY, Raji I, et al. Enhancing the immunogenicity of lipid-nanoparticle mRNA vaccines by adjuvanting the ionizable lipid and the mRNA. Nat Biomed Eng. 2023. doi:10.1038/s41551-023-01082-6
12. Marlin R, Godot V, Cardinaud S, et al. Targeting SARS-CoV-2 receptor-binding domain to cells expressing CD40 improves protection to infection in convalescent macaques. Nat Commun. 2021;12(1):5215. doi:10.1038/s41467-021-25382-0
13. Pastor Y, Ghazzaui N, Hammoudi A, Centlivre M, Cardinaud S, Levy Y. Refining the DC-targeting vaccination for preventing emerging infectious diseases. Front Immunol. 2022;13:949779. doi:10.3389/fimmu.2022.949779
14. Graham JP, Authie P, Yu CI, et al. Targeting dendritic cells in humanized mice receiving adoptive T cells via monoclonal antibodies fused to Flu epitopes. Vaccine. 2016;34(41):4857–4865. doi:10.1016/j.vaccine.2016.08.071
15. Liu Z, Xu W, Xia S, et al. RBD-Fc-based COVID-19 vaccine candidate induces highly potent SARS-CoV-2 neutralizing antibody response. Signal Transduct Target Ther. 2020;5(1):282. doi:10.1038/s41392-020-00402-5
16. Sun S, He L, Zhao Z, et al. Recombinant vaccine containing an RBD-Fc fusion induced protection against SARS-CoV-2 in nonhuman primates and mice. Cell Mol Immunol. 2021;18(4):1070–1073. doi:10.1038/s41423-021-00658-z
17. Kudriavtsev AV, Vakhrusheva AV, Kryuchkov NA, et al. Safety and immunogenicity of Betuvax-CoV-2, an RBD-Fc-Based SARS-CoV-2 recombinant vaccine: preliminary results of the first-in-human, randomized, double-blind, placebo-controlled Phase I/II clinical trial. Vaccines. 2023;11(2). doi:10.3390/vaccines11020326
18. Levin D, Golding B, Strome SE, Sauna ZE. Fc fusion as a platform technology: potential for modulating immunogenicity. Trends Biotechnol. 2015;33(1):27–34. doi:10.1016/j.tibtech.2014.11.001
19. Patel PN, Dickey TH, Hopp CS, et al. Neutralizing and interfering human antibodies define the structural and mechanistic basis for antigenic diversion. Nat Commun. 2022;13(1):5888. doi:10.1038/s41467-022-33336-3
20. Kinoshita T, Thyphronitis G, Tsokos GC, et al. Characterization of murine complement receptor type 2 and its immunological cross-reactivity with type 1 receptor. Int Immunol. 1990;2(7):651–659. doi:10.1093/intimm/2.7.651
21. Helmy KY, Katschke KJ, Gorgani NN, et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell. 2006;124(5):915–927. doi:10.1016/j.cell.2005.12.039
22. Schmidt RE, Gessner JE. Fc receptors and their interaction with complement in autoimmunity. Immunol Lett. 2005;100(1):56–67. doi:10.1016/j.imlet.2005.06.022
23. Erdei A, Isaák A, Török K, et al. Expression and role of CR1 and CR2 on B and T lymphocytes under physiological and autoimmune conditions. Mol Immunol. 2009;46(14):2767–2773. doi:10.1016/j.molimm.2009.05.181
24. Ahearn JM, Fischer MB, Croix D, et al. Disruption of the Cr2 locus results in a reduction in B-1a cells and in an impaired B cell response to T-dependent antigen. Immunity. 1996;4(3):251–262. doi:10.1016/s1074-7613(00)80433-1
25. Mandel TE, Phipps RP, Abbot A, Tew JG. The follicular dendritic cell: long term antigen retention during immunity. Immunol Rev. 1980;53:29–59. doi:10.1111/j.1600-065x.1980.tb01039.x
26. Ruffin N, Gea-Mallorquí E. Dynamics of antigen retention by follicular dendritic cells. Nat Rev Immunol. 2022;22(12):715. doi:10.1038/s41577-022-00798-x
27. Becherer JD, Lambris JD. Identification of the C3b receptor-binding domain in third component of complement. J Biol Chem. 1988;263(28):14586–14591.
28. Alsenz J, Becherer JD, Nilsson B, Lambris JD. Structural and functional analysis of C3 using monoclonal antibodies. Curr Top Microbiol Immunol. 1990;153:235–248. doi:10.1007/978-3-642-74977-3_13
29. Fishelson Z. Complement C3: a molecular mosaic of binding sites. Mol Immunol. 1991;28(4–5):545–552. doi:10.1016/0161-5890(91)90169-k
30. Oran AE, Isenman DE. Identification of residues within the 727–767 segment of human complement component C3 important for its interaction with factor H and with complement receptor 1 (CR1, CD35). J Biol Chem. 1999;274(8):5120–5130. doi:10.1074/jbc.274.8.5120
31. Hassett KJ, Benenato KE, Jacquinet E, et al. Optimization of lipid nanoparticles for intramuscular administration of mRNA vaccines. Mol Ther Nucleic Acids. 2019;15:1–11. doi:10.1016/j.omtn.2019.01.013
32. Schoenmaker L, Witzigmann D, Kulkarni JA, et al. mRNA-lipid nanoparticle COVID-19 vaccines: structure and stability. Int J Pharm. 2021;601:120586. doi:10.1016/j.ijpharm.2021.120586
33. Packer M, Gyawali D, Yerabolu R, Schariter J, White P. A novel mechanism for the loss of mRNA activity in lipid nanoparticle delivery systems. Nat Commun. 2021;12(1):6777. doi:10.1038/s41467-021-26926-0
34. Shihan MH, Novo SG, Le Marchand SJ, Wang Y, Duncan MK. A simple method for quantitating confocal fluorescent images. Biochem Biophys Rep. 2021;25:100916. doi:10.1016/j.bbrep.2021.100916
35. Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224(3):213–232. doi:10.1111/j.1365-2818.2006.01706.x
36. Nance KD, Meier JL. Modifications in an emergency: the role of N1-methylpseudouridine in COVID-19 vaccines. ACS Cent Sci. 2021;7(5):748–756. doi:10.1021/acscentsci.1c00197
37. Granados-Riveron JT, Aquino-Jarquin G. Engineering of the current nucleoside-modified mRNA-LNP vaccines against SARS-CoV-2. Biomed Pharmacother. 2021;142:111953. doi:10.1016/j.biopha.2021.111953
38. Xia X. Detailed dissection and critical evaluation of the Pfizer/BioNTech and moderna mRNA vaccines. Vaccines. 2021;9(7). doi:10.3390/vaccines9070734
39. Trepotec Z, Geiger J, Plank C, Aneja MK, Rudolph C. Segmented poly(A) tails significantly reduce recombination of plasmid DNA without affecting mRNA translation efficiency or half-life. RNA. 2019;25(4):507–518. doi:10.1261/rna.069286.118
40. Xiang K, Bartel DP. The molecular basis of coupling between poly(A)-tail length and translational efficiency. Elife. 2021;10. doi:10.7554/eLife.66493
41. Dussupt V, Sankhala RS, Mendez-Rivera L, et al. Low-dose in vivo protection and neutralization across SARS-CoV-2 variants by monoclonal antibody combinations. Nat Immunol. 2021;22(12):1503–1514. doi:10.1038/s41590-021-01068-z
42. Shang J, Wan Y, Luo C, et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci U S A. 2020;117(21):11727–11734. doi:10.1073/pnas.2003138117
43. Corti D, Purcell LA, Snell G, Veesler D. Tackling COVID-19 with neutralizing monoclonal antibodies. Cell. 2021;184(17):4593–4595. doi:10.1016/j.cell.2021.07.027
44. Harvey WT, Carabelli AM, Jackson B, et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol. 2021;19(7):409–424. doi:10.1038/s41579-021-00573-0
45. Liu Y, Soh WT, Kishikawa JI, et al. An infectivity-enhancing site on the SARS-CoV-2 spike protein targeted by antibodies. Cell. 2021;184(13):3452–3466.e18. doi:10.1016/j.cell.2021.05.032
46. Zang J, Gu C, Zhou B, et al. Immunization with the receptor-binding domain of SARS-CoV-2 elicits antibodies cross-neutralizing SARS-CoV-2 and SARS-CoV without antibody-dependent enhancement. Cell Discov. 2020;6:61. doi:10.1038/s41421-020-00199-1
47. Sun S, Cai Y, Song TZ, et al. Interferon-armed RBD dimer enhances the immunogenicity of RBD for sterilizing immunity against SARS-CoV-2. Cell Res. 2021;31(9):1011–1023. doi:10.1038/s41422-021-00531-8
48. Xia S, Duan K, Zhang Y, et al. Effect of an inactivated vaccine against SARS-CoV-2 on safety and immunogenicity outcomes: interim analysis of 2 randomized clinical trials. JAMA. 2020;324(10):951–960. doi:10.1001/jama.2020.15543
49. Liu Z, Zhou J, Xu W, et al. A novel STING agonist-adjuvanted pan-sarbecovirus vaccine elicits potent and durable neutralizing antibody and T cell responses in mice, rabbits and NHPs. Cell Res. 2022;32(3):269–287. doi:10.1038/s41422-022-00612-2
50. Du L, Zhao G, He Y, et al. Receptor-binding domain of SARS-CoV spike protein induces long-term protective immunity in an animal model. Vaccine. 2007;25(15):2832–2838. doi:10.1016/j.vaccine.2006.10.031
51. Brandys P, Montagutelli X, Merenkova I, et al. A mRNA vaccine encoding for a RBD 60-mer nanoparticle elicits neutralizing antibodies and protective immunity against the SARS-CoV-2 delta variant in transgenic K18-hACE2 mice. Front Immunol. 2022;13:912898. doi:10.3389/fimmu.2022.912898
52. Tai W, Zhang X, Drelich A, et al. A novel receptor-binding domain (RBD)-based mRNA vaccine against SARS-CoV-2. Cell Res. 2020;30(10):932–935. doi:10.1038/s41422-020-0387-5
53. Kontermann RE. Strategies for extended serum half-life of protein therapeutics. Curr Opin Biotechnol. 2011;22(6):868–876. doi:10.1016/j.copbio.2011.06.012
54. Uthaipibull C, Aufiero B, Syed SE, et al. Inhibitory and blocking monoclonal antibody epitopes on merozoite surface protein 1 of the malaria parasite Plasmodium falciparum. J Mol Biol. 2001;307(5):1381–1394. doi:10.1006/jmbi.2001.4574
55. Ross TM, Xu Y, Bright RA, Robinson HL. C3d enhancement of antibodies to hemagglutinin accelerates protection against influenza virus challenge. Nat Immunol. 2000;1(2):127–131. doi:10.1038/77802
56. Green TD, Newton BR, Rota PA, Xu Y, Robinson HL, Ross TM. C3d enhancement of neutralizing antibodies to measles hemagglutinin. Vaccine. 2001;20(1–2):242–248. doi:10.1016/s0264-410x(01)00266-3
57. Bower JF, Sanders KL, Ross TM. C3d enhances immune responses using low doses of DNA expressing the HIV-1 envelope from codon-optimized gene sequences. Curr HIV Res. 2005;3(2):191–198. doi:10.2174/1570162053506937
58. Fang Y, Xu C, Fu YX, Holers VM, Molina H. Expression of complement receptors 1 and 2 on follicular dendritic cells is necessary for the generation of a strong antigen-specific IgG response. J Immunol. 1998;160(11):5273–5279.
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.