")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 11
Breaking the Barriers of Therapy Resistance: Harnessing Ferroptosis for Effective Hepatocellular Carcinoma Therapy
Authors Lv X, Lan G, Zhu L , Guo Q
Received 18 March 2024
Accepted for publication 11 June 2024
Published 2 July 2024 Volume 2024:11 Pages 1265—1278
DOI https://doi.org/10.2147/JHC.S469449
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr David Gerber
Xianmei Lv,1,2,* Gaochen Lan,3,* Lujian Zhu,4,* Qiusheng Guo1
1Department of Radiotherapy, Jinhua People’s Hospital, Jinhua, Zhejiang, 321000, People’s Republic of China; 2Department of Oncology, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, 362000, People’s Republic of China; 3Department of Infectious Diseases, Affiliated Jinhua Hospital, Zhejiang University School of Medicine, Jinhua, Zhejiang, 321000, People’s Republic of China; 4Department of Medical Oncology, Affiliated Jinhua Hospital, Zhejiang University School of Medicine, Jinhua, Zhejiang, 321000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qiusheng Guo, Department of Medical Oncology, Affiliated Jinhua Hospital, Zhejiang University School of Medicine, Jinhua, Zhejiang, 321000, People’s Republic of China, Email [email protected]
Abstract: Ferroptosis is a type of cell death that relies on iron and is distinguished by the occurrence of lipid peroxidation and the buildup of reactive oxygen species. Ferroptosis has been demonstrated to have a significant impact on the advancement and resistance to treatment of hepatocellular carcinoma (HCC), thereby highlighting its potential as a viable therapeutic target. Ferroptosis was observed in HCC tissues in contrast to normal liver tissue. The inhibition of ferroptosis has been found to increase the viability of HCC cells and decrease their susceptibility to various anticancer therapies, including chemotherapy, radiotherapy, and immune checkpoint blockade. The administration of drugs that directly modulate ferroptosis regulators or induce excessive production of lipid-reactive oxygen species has demonstrated the potential to enhance the responsiveness of drug-resistant HCC cells to treatment. However, the precise mechanism underlying this phenomenon remains ambiguous. This review presents a comprehensive overview of the crucial role played by ferroptosis in enhancing the efficacy of treatment for hepatocellular carcinoma (HCC). The main aim of this study is to examine the feasibility of utilizing ferroptosis as a therapeutic approach to improve the efficacy of HCC treatment and overcome drug resistance.
Keywords: ferroptosis, hepatocellular carcinoma, chemotherapy, tyrosine kinase inhibitor, immunosuppressive therapy, radiotherapy
Graphical Abstract:
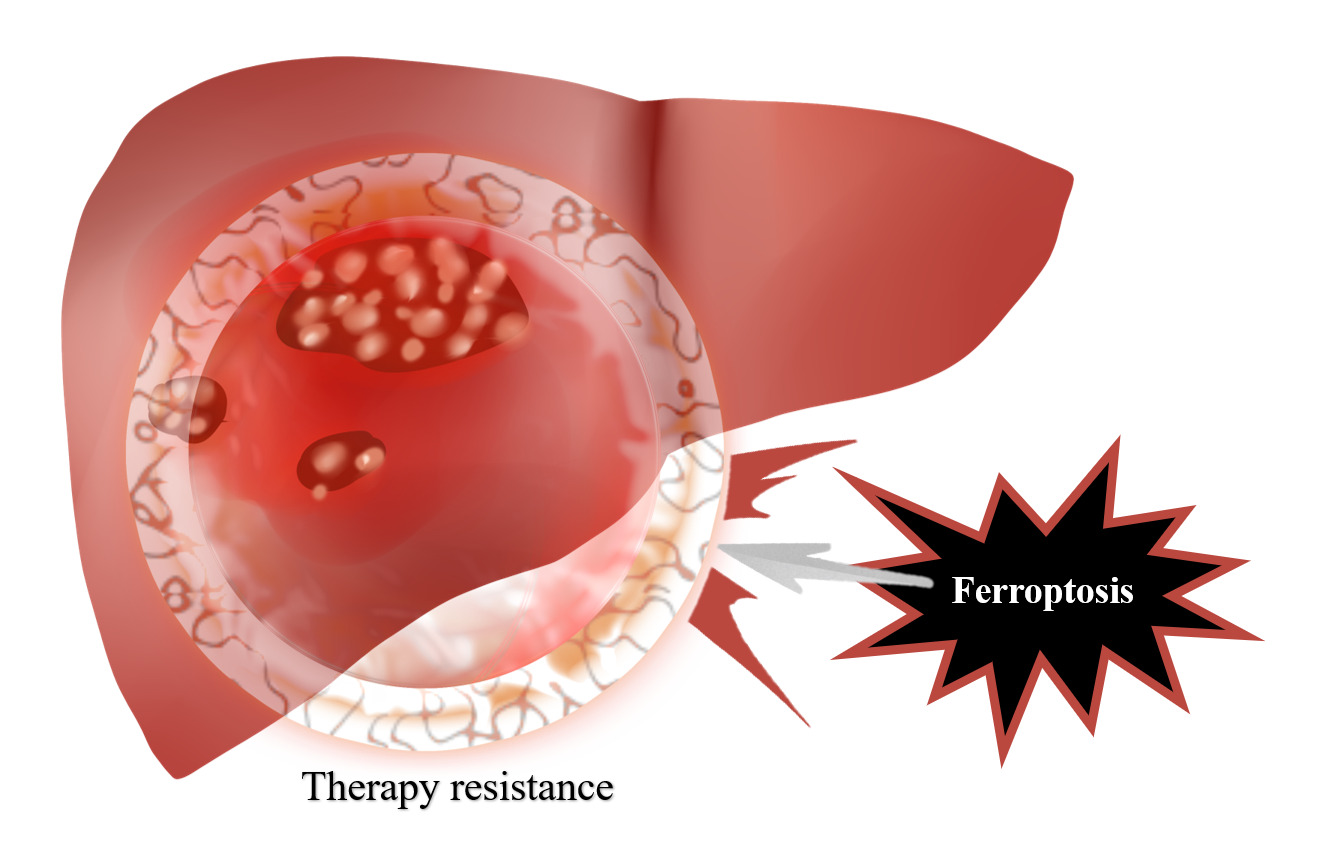
Introduction
Hepatocellular carcinoma (HCC) is a prevalent malignant neoplasm that presents a significant risk to human well-being and survival. In recent years, there has been an observed increase in the occurrence and fatality rates of HCC. It was projected that the annual incidence of HCC and HCC-related mortality could rise by approximately 500,000 by 2040.1 This alarming trend could only be reversed through the implementation of effective primary prevention. The therapeutic approaches for HCC typically encompass a range of interventions such as surgical resection, ablation techniques, embolization procedures, radiotherapy, molecular targeted therapy, immunotherapy, and chemotherapy. These strategies have been extensively studied and documented in the literature.2 Despite some advancements in the diagnosis and treatment of HCC, the overall prognosis for patients with HCC remained unfavorable (https://www.cancer.org/). The high resistance of HCC to different treatment modalities, such as chemotherapy, has been extensively documented.3 HCC cells could reduce their responsiveness to therapeutic interventions through a range of mechanisms, including mitophagy,4 epigenetic regulation,5 and ATP binding cassette (ABC) transporters.6 In addition, previous studies have shown that various factors in the tumor microenvironment, such as hypoxia7 and immune infiltration, played a significant role in promoting the development of drug resistance in HCC.8 One of the primary challenges in the clinical management of HCC is the effective enhancement of treatment sensitivity.
Iron is a crucial trace element for organisms; however, an excessive amount of iron can lead to cytotoxicity through the catalysis of reactive oxygen species (ROS) production. Ferroptosis is a type of cell death that is dependent on iron and is characterized by the process of lipid peroxidation.9 During the process of ferroptosis, the buildup of intracellular iron ions leads to the production of highly reactive hydroxyl radicals through the Fenton reaction. This, in turn, causes irreversible lipid peroxidation. The aforementioned process results in the degradation of the integrity and functionality of cellular membranes, as well as the impairment of DNA and other essential molecules, ultimately leading to the demise of the cell. The dysregulation of iron homeostasis,10,11 aberrant lipid metabolism,12 and ROS accumulation13 contributed to the heightened vulnerability of HCC cells to ferroptosis. Direct modulation of ferroptotic regulators has been demonstrated to restore the sensitivity of HCC cells to chemotherapy.14 Therefore, the utilization of ferroptosis as a strategy to overcome chemoresistance in HCC has the potential to enhance the overall survival rates of HCC patients.
In this review, we aim to provide a comprehensive summary of the existing evidence derived from both preclinical and clinical studies, focusing on the significance of ferroptosis in conferring survival advantages and therapy resistance to HCC cells. Specifically, we will explore the impact of ferroptosis on HCC cells’ response to different treatment modalities, such as radiotherapy and chemotherapy. This review aims to provide a comprehensive analysis of the role of ferroptosis in drug resistance in HCC, to identify novel therapeutic targets to overcome resistance. This study aims to elucidate the potential application and limitations of ferroptosis-based strategies in reversing drug resistance in HCC. The findings of this research will serve as a valuable reference for future basic and clinical investigations in this field.
Causes Contributing to Diminished Treatment Efficacy in HCC
Multiple factors in liver cancer cells and tumor microenvironment, including DNA damage repair, immunosuppression, drug transport, and metabolic reprogramming, together lead to reduced sensitivity to liver cancer therapy (Figure 1).
 |
Figure 1 Factors contributing to diminished treatment efficacy in hepatocellular carcinoma (HCC). Abbreviations: METTL1, Methyltransferase Like 1; BRCA1, BReast CAncer gene 1; TonEBP, Tonicity-responsive Enhancer Binding Protein; XRCC1, X-Ray Repair Cross Complementing 1; HCC, Hepatocellular Carcinoma; TAM, Tumor-Associated Macrophage; Treg, Regulatory T cell; ABC, ATP-Binding Cassette; P-gp, P-glycoprotein; ALDH, ALdehyde DeHydrogenase; UGT, UDP-GlucuronosylTransferase; CYP, CYtochrome P450; mTOR, mechanistic Target Of Rapamycin; HIF1α, Hypoxia-Inducible Factor 1-alpha; SREBP1, Sterol Regulatory Element-Binding Protein 1; GLUT1, Glucose Transporter 1; PFK1, Phosphofructokinase 1; CRLLS1, Cardiolipin Synthase 1; UBQLN1, Ubiquilin 1; PGC1β, Peroxisome proliferator-activated receptor Gamma Coactivator 1-beta; G6PC, Glucose-6-Phosphatase Catalytic subunit. |
The DNA Mutations and Repair Capacity of HCC Cells Contribute to Their Resistance to Treatment
The modifications in the DNA mutation and repair capacity within HCC cells contribute to their resistance to therapeutic interventions. The DNA mutations observed in HCC cells encompass a range of forms, such as gene mutations, chromosomal abnormalities, DNA methylation, and nucleotide substitutions. These mutations had the potential to impact essential cellular processes such as growth, division, apoptosis, and DNA repair.15 Key genes involved in DNA repair, such as TP53, BRCA1, and XRCC1, were commonly found to be mutated in HCC cells. These mutations contribute to the development of resistance to radiotherapy and chemotherapy.16–19 In the field of radiotherapy, HCC cells were exposed to radiation energy, which induces both single- and double-strand breaks in the DNA. As a result, these breaks led to either apoptosis or a cessation of halted proliferation.20 However, mutations in the DNA of HCC cells potentially hindered the effectiveness of DNA repair mechanisms, resulting in reduced sensitivity to radiotherapy and diminished therapeutic outcomes. It has been discovered that the aberrant functioning of the RAD21 DNA repair pathway in HCC cells contributed to a decrease in sensitivity to radiotherapy. Overexpression of RAD21 has been shown to result in a reduction in the concentration of 8-hydroxy-2-deoxyguanosine in the medium, as well as a decrease in the levels of γH2AX and ATM proteins. These findings suggest that there is a mitigation of DNA damage in radiated HCC cells.21 Furthermore, the identification of an enzyme called METTL1 has revealed its significant involvement in the radioresistance of HCC cells. METTL1 enhanced the repair process of DNA double-strand breaks, thereby providing HCC cells with resistance against ionizing radiation. Methyltransferase-mediated modifications of tRNA have been shown to have a selective regulatory effect on the translation of specific essential proteins, thereby increasing the efficiency of non-homologous end-joining repair.22 In the context of chemotherapy, HCC cells changed their growth and proliferation status as a result of exposure to chemotherapeutic agents. However, DNA mutations in HCC cells could lead to alterations or deficiencies in crucial genes, thereby compromising their ability to respond to chemotherapeutic agents and reducing the effectiveness of treatment.23 For example, mutations in the p53 gene have been found to contribute to the development of resistance to arsenic trioxide and an increased ability of HCC cells to metastasize. Silencing the expression of mutant p53 restored sensitivity to arsenic trioxide in resistant cells and suppressed metastatic activities. Conversely, overexpression of mutant p53 had the opposite effects.24 In HCC, the tumor suppressor protein p53 played a crucial role in regulating ferroptosis.25 This suggested that ferroptosis could serve as a significant regulatory mechanism for cells to overcome drug resistance. Cisplatin elicited the recruitment of the ERCC1/XPF dimer to chromatin in a manner that was dependent on nucleotide excision repair. This process ultimately led to DNA repair and the development of resistance to cisplatin.26
In summary, the alterations in DNA mutations and repair capabilities were major factors contributing to HCC cell resistance to therapies. Further elucidating the relationship between aberrant DNA repair mechanisms and drug resistance in HCC cells will facilitate the discovery of new therapeutic approaches and targeted drugs to improve HCC treatment efficacy.
The Microenvironment of HCC Created an Immunosuppressive Niche to Evade Therapeutic Interventions
The HCC microenvironment encompassed the intricate interplay and reciprocal influences between the surrounding tissues and cells to HCC cells. Multiple mechanisms are present in the HCC microenvironment that contribute to reduced responsiveness to radiotherapy and chemotherapy. These mechanisms include immune evasion, the presence of HCC stem cells, and hypoxia.27 Spatial transcriptomic analysis has provided insights into the composition of the HCC microenvironment, demonstrating an enrichment of immune cells and cancer-associated fibroblasts. Furthermore, this microenvironment exhibited the expression of immune desert signatures and cancer stem cell markers, which have been associated with chemoresistance.28 The HCC microenvironment is characterized by the presence of numerous immunosuppressive cells and factors, such as tumor-associated macrophages (TAMs), regulatory T cells (Treg cells), and anti-inflammatory cytokines. TAMs exhibited immunosuppressive properties, which served to inhibit the functions of activated immune cells. In the microenvironment of HCC, TAMs often demonstrated a heightened level of immunosuppressive activity. Treg cells possessed the ability to suppress other immune cells. In HCC, an increase in Treg cell levels led to a dampening of immune responses. Additionally, the microenvironment of HCC was characterized by elevated levels of anti-inflammatory cytokines such as IL-10 and TGF-β. These cytokines could suppress immune cell function and compromise the effectiveness of anti-tumor immune responses. The existence of these immune evasion mechanisms resulted in reduced sensitivity of HCC cells to radiotherapy and chemotherapy, thereby restricting the enhancement of therapeutic effectiveness.29,30 The presence and function of HCC stem cells were sustained by the tumor microenvironment.31 HCC stem cells represented a distinct and limited subset within HCC tissues, demonstrating elevated levels of drug resistance and aggressiveness in comparison to conventional HCC cells. The FZD10-β-catenin/c-Jun axis in HCC stem cells played a crucial role in the transcriptional regulation of METTL3 expression through a positive feedback loop, thereby imparting resistance to sorafenib treatment.32 Furthermore, the hypoxic microenvironment in HCC played a significant role in diminishing treatment efficacy. Under conditions of hypoxia, the nuclear translocation of PTPN14 overexpressed downstream YAP, leading to the development of resistance to sorafenib.33 Concurrently, the HSP90α protein interacted with the necrosome complex and facilitated chaperone-mediated autophagy degradation, thereby inhibiting programmed necrotic cell death.34 Hypoxia has been found to induce resistance to radiotherapy in HCC cells. This resistance was mediated through the activation of the PDK1 signaling pathway, which led to enhanced cell motility and expression of stemness markers. Additionally, hypoxia impairs DNA repair mechanisms and reduces the radiosensitivity of HCC cells.35
Drug Metabolism Processes Influence Therapeutic Efficacy in HCC
The drug metabolism processes, encompassing absorption, distribution, metabolism, and excretion (ADME), are pivotal in influencing the therapeutic effectiveness in HCC. Disruptions in these processes may result in decreased drug levels within HCC cells, consequently diminishing the efficacy of therapeutic strategies in inhibiting tumor proliferation.36 Genetic variations in ADME-related genes have been linked to differences in the effectiveness of sorafenib. A study identified specific single nucleotide polymorphisms in genes such as ADH1A, ADH6, SULT1A2/CCDC101, CYP26A1, DPYD, FMO2, and SLC22A14 that were significantly associated with the response to sorafenib in patients with HCC.37 These results indicate that individual variances in ADME processes may play a critical role in the variability observed in sorafenib efficacy.
The physicochemical properties of sorafenib, including its limited water solubility and rapid clearance, contribute to its low oral absorption efficiency.38,39 These attributes constrain its clinical utility and hinder the expression of its antitumor properties. The spatial heterogeneity of HCC tissues may influence the intra-tumoral distribution of therapeutic agents. The co-administration of bufalin and sorafenib resulted in a reduction of spatial heterogeneity within the HCC-LM3 model, indicating a potential enhancement of therapeutic efficacy through the promotion of uniform drug distribution.40 Additionally, circulating tumor cells have been shown to establish a protective barrier through the adherence to and activation of platelets, leading to the formation of microthrombi that impede the effective eradication of CTCs by therapeutic agents and immune cells.41
Several studies have reported that the expression of P-gp and multidrug resistance protein 2 could modulate the genomic effects of estrogen receptors and pregnane X receptors, thereby playing a role in the regulation of drug transport proteins and facilitating the development of resistance to sorafenib.42 The overexpression of multidrug resistance protein 2 has been found to confer resistance to both 5-FU and cisplatin.43 Intervening in the expression of breast cancer resistance protein using RNA interference technology has been shown to enhance the sensitivity of the Hep G2 cell line to doxorubicin.44
In terms of drug metabolism, the enzymes aldehyde dehydrogenase45 and UDP-glucuronosyltransferase46 have been identified as key factors contributing to the development of sorafenib resistance in HCC. The downregulation of cytochrome P450 (CYP) proteins in HCC is associated with a significant decrease in CYP activity, with a strong correlation observed between CYP protein levels and activity. CYP1A2 and CYP2B6 enzymes play a role in the metabolism of various drugs like linivanil, banoxantrone, exemestane, voreloxin, and cyclophosphamide.47–49
Metabolic Reprogramming in Hepatocellular Carcinoma Led to Decreased Sensitivity to Treatment
In the progression of HCC, neoplastic cells underwent metabolic reprogramming to accommodate their accelerated growth and proliferation. This metabolic remodeling involved an upregulation of glycolysis and fatty acid synthesis, accompanied by a downregulation of processes such as the tricarboxylic acid cycle and oxidative phosphorylation.50 As a result of this remodeling process, HCC cells demonstrated diminished responsiveness to specific conventional therapeutic approaches. The application of the glycolysis inhibitor 2-deoxyglucose has been shown to reduce cell viability in sorafenib-resistant HCC cells, impede colony formation, and induce G0/G1 cell cycle arrest. Consequently, this significantly augmented the cytotoxic impact of sorafenib.51 HCC cells that are resistant to radiation therapy demonstrated a significant reliance on glucose. These cells utilized glucose to facilitate the synthesis of phospholipids instead of the atypical glycolytic pathway that produced lactate and pyruvate while inhibiting apoptosis. This physiological process was governed by various signaling molecules, including mTORC1, HIF-1α, and SREBP1.52 Upregulation of UBQLN1 in sorafenib-resistant HCC cells has been shown to facilitate the ubiquitin-independent degradation of PGC1β protein, leading to the remodeling of mitochondrial and redox metabolism.53 The miR-494/G6pc axis has been identified as a key factor in the metabolic plasticity of tumor cells, specifically in promoting the metabolic shift towards a glycolytic phenotype in HCC cells. This metabolic adaptation enabled the survival of HCC cells under cytotoxic conditions induced by sorafenib treatment.54
Furthermore, alongside changes in glucose and lipid metabolism, HCC cells demonstrate notable disruptions in iron metabolism when compared to healthy hepatocytes. During the progression of hepatocellular carcinoma, tumor cells undergo reprogramming of iron homeostasis to meet the increased demand for iron in rapidly dividing cancer cells. This reprogramming involves the upregulation of membrane iron transporters, such as transferrin receptor and divalent metal transporter 1, which enhance the uptake of plasma iron into tumor cells.55,56 Additionally, the downregulation of hepcidin expression in hepatocytes located within tumor tissues decreases intracellular iron transport to plasma, disrupting the body’s regulation of iron levels.57 The modulation of proteins involved in intracellular iron storage and utilization undergoes alterations in liver cancer. Specifically, the upregulation of Ferritin light chain in liver cancer tissues is associated with tumor grade and prognosis.58 The upregulation of mitochondrial iron transporter facilitates iron accumulation in mitochondria, thereby ensuring an adequate supply of iron for electron transport in the respiratory chain and DNA synthesis.59 Meanwhile, the upregulation of the iron death inhibitor (FDFT1) is commonly observed in liver cancer tissues, inhibiting iron-dependent cell death and sustaining elevated levels of intracellular free iron.60 These modifications in iron metabolism collectively result in markedly increased iron levels in liver cancer tissues when compared to adjacent non-cancerous tissues and normal liver tissues.
Targeting Ferroptosis as a Sensitization Strategy for the Treatment of HCC
Chemotherapy
Tumor cells utilized diverse mechanisms to hinder ferroptosis that is triggered by chemotherapy drugs. These mechanisms encompassed various regulatory processes, such as miRNA-mediated regulation influenced by factors present in the tumor microenvironment, modification of key metabolic enzymes and non-coding RNAs to alter gene expression, and the direct inhibition of ferroptosis by proteins associated with tumors. Tumor cells could inhibit ferroptosis, which is induced by chemotherapy drugs, through the release of small RNAs. For instance, in pancreatic cancer, fibroblasts hindered the expression of acyl-CoA synthetase long-chain family member 4 (ACSL4). This inhibition was achieved through the release of extracellular vesicles that contain miR-3173-5p. Consequently, the ferroptosis induced by gemcitabine is attenuated.61 Abnormal expression and functionality of specific metabolic enzymes and non-coding RNAs had the potential to influence ferroptosis triggered by chemotherapy drugs. For example, Acyl-CoA dehydrogenase medium chain mitigated lipid peroxidation and ferroptosis caused by chemotherapy drugs through the enhancement of unsaturated fatty acid metabolism.62 DACT3 antisense RNA 1, hypoxia-induced long non-coding RNA (LncRNA), also participated in the development of resistance through SIRT1-mediated ferroptosis.63 The upregulation of LINC01134 has been found to augment the cytotoxic efficacy of the chemotherapeutic agent oxaliplatin through the inhibition of Glutathione peroxidase 4 (GPX4).64 Certain proteins associated with tumors could directly impede ferroptosis that is triggered by chemotherapy drugs. Phosphorylated lysyl oxidase-like 3 has been found to stabilize DHODH, thereby exerting an inhibitory effect on ferroptosis induced by chemotherapy.65 Elevated expression of ferritin has been found to confer resistance to ferroptosis in tumor cells.66 In hepatitis B virus-related HCC, it was important to consider the influence of the virus on pathways of cell death that are dependent on iron. Hepatitis B virus infection decreased intracellular Fe2+ levels and induced the expression of GPX4 through the Serine and arginine-rich splicing factor 2 / PCNA clamp associated factor tv1 axis. This mechanism ultimately resulted in a reduction of ferroptosis and contributed to the development of sorafenib resistance.67 Further clarification of the molecular mechanisms that underlie the inhibition of ferroptosis will contribute to the advancement of inhibitors that target these proteins, ultimately improving the sensitivity of chemotherapy.
Meanwhile, chemotherapeutic agents induced ferroptosis and enhance their cytotoxicity or that of other drugs. For instance, the administration of doxorubicin elevated ROS levels, thereby inducing ferroptosis.68 Boric acid administration resulted in an elevation of intracellular reactive oxygen species, glutathione, and thiobarbituric acid reactive substances levels. Consequently, it potentiated the occurrence of ferroptosis, which was induced by RAS-selective lethal 3.69 Rationally designed combination chemotherapy regimens had the potential to induce ferroptosis by leveraging synergistic effects, thereby enhancing treatment sensitivity. The concurrent administration of gimatecan and cisplatin resulted in the induction of lipid peroxidation, which led to the accumulation of iron and the subsequent initiation of ferroptosis in malignant cells.70 Some drugs that are commonly used in clinical settings could induce ferroptosis either independently or in combination, thereby offering additional possibilities for cancer chemotherapy. For example, Caragliorubicin was discovered to induce ferroptosis through the modulation of glycolysis and glutamine metabolism, thereby increasing the sensitivity of tumor cells to cisplatin.71 This study presented a range of clinically approved drug options that can be utilized in combination to enhance the effectiveness of therapy.
In conclusion, the elucidation of the fundamental intracellular and extracellular molecular mechanisms that regulate ferroptosis holds the potential to enhance the sensitivity of tumor chemotherapy (Figure 2). By targeting these specific nodes, a deeper understanding of the comprehensive treatment of cancer can be achieved, leading to novel insights in this field.
 |
Figure 2 HCC resistance to chemotherapy drugs is regulated by ferroptosis. By figdraw. Abbreviations: GSH, Glutathione; GS-SH, Glutathione (reduced form); GPX4, Glutathione Peroxidase 4; Nrf2, Nuclear factor erythroid 2-related factor 2; PUFAs-OH, Polyunsaturated Fatty Acids-Hydroxyl; PUFAs-OO, Polyunsaturated Fatty Acids-Peroxyl; ROS, Reactive Oxygen Species; CoQH2, Coenzyme Q10 (reduced form); CoQ, Coenzyme Q10; DHODH, Dihydroorotate Dehydrogenase; AK2, Adenylate Kinase 2; LOXL3, Lysyl Oxidase Like 3; NCOA4, Nuclear Receptor Coactivator 4; PCLAF tv1, PCLAF transcript variant 1; SRSF2, Serine and Arginine Rich Splicing Factor 2; HBV, Hepatitis B Virus; IREB2, Iron Responsive Element Binding Protein 2; DMTI, Divalent Metal Transporter 1; STEAP3, Six Transmembrane Epithelial Antigen of Prostate 3; FTH, Ferritin Heavy chain; TFR, Transferrin Receptor. |
Tyrosine Kinase Inhibitor
Sorafenib, a broad-spectrum tyrosine kinase inhibitor, was the first systemic drug to receive FDA approval for the first-line treatment of HCC.72 Currently, sorafenib has gained significant popularity in clinical practice as a targeted therapy for advanced HCC, and it plays a pivotal role in the treatment of HCC. However, the therapeutic outcomes in patients treated with sorafenib are not very favorable. The overall survival rate of patients treated with sorafenib does not surpass 3 years. Additionally, 11% of patients experienced severe treatment-related adverse events.73 The prevalence of sorafenib resistance in HCC is becoming increasingly common.74
Sorafenib has been shown to induce ferroptosis through multiple pathways, including the promotion of lipid peroxidation, inhibition of glutathione synthesis, reduction of glutathione levels, and suppression of nuclear factor erythroid 2-related factor 2 (Nrf2) signaling.25,75–77 Activation of Nrf2 has been found to enhance cancer cell survival and proliferation, induce changes in cancer metabolism, and influence the immune response within the tumor microenvironment, ultimately leading to the resistance to sorafenib.78 A study conducted by Sun et al also demonstrated that the Nrf2 pathway suppresses sorafenib-induced ferroptosis by upregulating metallothionein −1G, leading to increased antioxidant capacity in HCC and subsequently diminishing the effectiveness of sorafenib treatment.79,80 HCC cells activated other pathways to counteract the induction of ferroptosis by sorafenib. Several pathways have been identified to play a role in inhibiting ferroptosis. For example, the signaling pathway involving the proto-oncogene tyrosine-protein kinase Src, p53, and stearoyl-CoA desaturase 1 inhibited lipid peroxidation, which is a crucial mechanism in the occurrence of ferroptosis.81 Additionally, Double Homeobox A Pseudogene 8 stabilized SLC7A11 expression, thereby suppressing ferroptosis.82 Oncoprotein hepatitis B X–interacting protein directly upregulated stearoyl-CoA desaturase 1 expression, leading to the inhibition of ferroptosis.83 Furthermore, Partner of NOB1 Homolog promoted glutathione synthesis, which helps in resisting ferroptosis.84
Targeted inhibition of the key molecules involved in the aforementioned resistance pathways has the potential to augment the ferroptotic effects of sorafenib. Targeting the SLC7A11 potentially enhanced the synergistic antitumor effects of sorafenib, a kinase inhibitor, and iron chelators.85 Inhibition of Partner of NOB1 Homolog decreased the synthesis of glutathione, thereby promoting ferroptosis.84 The enhancement of ferroptosis in HCC cells could be achieved by either suppressing glutathione synthesis or increasing glutathione consumption. Copper disulfide depleted glutathione, thereby inducing ferroptosis.86 Nrf2 inhibitors have been shown to enhance the cytotoxic effects of sorafenib.77 Inhibiting the transcription factor Nrf2 or its downstream genes, such as GPX4, has emerged as a potential approach to enhance the effectiveness of sorafenib treatment.76,87 Meanwhile, certain agents induced or enhanced ferroptosis in HCC cells, thereby increasing the sensitivity to sorafenib. Examples of such agents included caragliorubicin,71 light-activated sorafenib complexes,88 and manganese silicon oxide nanoparticles.89 Copper disulfide complexes have been found to possess the ability to induce ferroptosis and can further augment this process through the inhibition of Nrf2 signaling, as reported in a recent study.86 Activating estrogen-related receptor γ elicited ferroptosis through the facilitation of mitochondrial ROS production, thereby augmenting the sensitivity of sorafenib treatment.90
Lenvatinib, another tyrosine kinase inhibitor utilized in the treatment of advanced HCC, has been observed to induce ferroptosis. Lenvatinib functions by inhibiting fibroblast growth factor receptor-4, thereby suppressing the Nrf2 pathway, increasing lipid peroxidation, and ultimately leading to ferroptosis.91 The expression level of FGFR4 has been linked to the effectiveness of Lenvatinib and the progression-free survival of patients with HCC. Additionally, lncRNA HAND2-AS1 can effectively overcome resistance to Lenvatinib by acting as a competitive inhibitor of miR-219a-1-3p, consequently upregulating the expression of ferroptosis-associated genes such as toll-like receptor 4, NADPH oxidase 2, and dual oxidase 2.92
In conclusion, the investigation of the precise mechanisms underlying HCC cell resistance to sorafenib-induced ferroptosis, as well as the development of targeted interventions targeting the crucial nodes within these mechanisms, offer novel avenues and strategies for enhancing the sensitivity of sorafenib in the treatment of HCC (Figure 3).
 |
Figure 3 Ferroptosis regulates tyrosine kinase inhibitor resistance in HCC. Abbreviations: SCD, Stearoyl-CoA Desaturase; HBXIP, Hepatitis B X–Interacting Protein; P53, Tumor Protein P53; URI, Unconventional prefoldin RPB5 Interactor; TRIM28, Tripartite Motif Containing 28; MDM2, Mouse Double Minute 2 homolog; PNO1, Partner of NOB1 homolog; MTIG, Metallothionein 1G; KEAP1, Kelch-like ECH-Associated Protein 1; FGFR4, Fibroblast Growth Factor Receptor 4. |
Immunosuppressive Therapy
Numerous inhibitory immune cells, including regulatory T cells (Tregs) and bone marrow-derived suppressor cells (MDSCs), are present in the microenvironment of liver cancer. Through the secretion of inhibitory cytokines such as IL-10 and TGF-β, these cells can prompt tumor cells to upregulate the immune checkpoint molecule programmed death 1 (PD-1), facilitating evasion of immune surveillance.93 Notably, elevated programmed death ligand 1 (PD-L1) expression in HCC cells is strongly correlated with poorer overall survival outcomes,94 underscoring the potential utility of PD-1/PD-L1 immune checkpoint inhibition therapy as a promising treatment strategy.
Recent research has further revealed a notable association between ferroptosis and the responsiveness of HCC to PD-1 therapy. Inducing ferroptosis in HCC cells could directly inhibit tumor progression. Inhibiting phosphoglycerate mutase 1 had antitumor effects by inducing ferroptosis in HCC cells and promoting the infiltration of CD8+ T cells. It could also decrease PD-L1 expression to improve the effectiveness of anti-PD-1 treatment, offering a new approach to treating HCC.95 Nanosized contrast agents carrying drugs induced immunogenic and ferroptosis cell death in HCC cells, activated immune responses in the tumor microenvironment, inhibited lung metastasis, and produced synergistic effects when combined with anti-PD-L1 treatment.96 High expression of mitochondrial translocator protein was linked to the progression of HCC and a negative prognosis. It activated the Nrf2 antioxidant signaling pathway to inhibit ferroptosis in HCC cells. Additionally, the upregulation of PD-L1 expression by translocator protein can hinder immune surveillance in HCC.97 The deletion of GPX4 did not inhibit hepatocellular carcinogenesis. Instead, it induced the expression of PD-L1 in tumor cells and promoted the infiltration of myeloid-derived suppressor cells to suppress immune responses. The co-administration of anti-PD-1 agents with ferroptosis-inducing drugs exhibited augmented efficacy in the treatment of HCC.98 Ferroptosis remodeled the tumor microenvironment to enhance the efficacy of PD-1 treatment. Selectively targeting xCT in TAMs could inhibit HCC growth and metastasis of HCC by inducing ferroptosis in TAMs. Additionally, it had the potential to enhance the upregulation of PD-L1 on TAMs, thereby resulting in synergistic outcomes when administered in conjunction with anti-PD-L1 therapy.99 A high expression of APOC1 was linked to M2 polarization of TAMs, while the induction of M1 polarization through the ferroptosis pathway was accomplished by suppressing apolipoprotein C1. This remodeling of the tumor microenvironment enhanced the efficacy of anti-PD-1 therapy.100
Hence, conducting a study on the association between ferroptosis and HCC immunotherapy has the potential to provide valuable insights into enhancing the effectiveness of PD-1 treatment in HCC. The aforementioned objective can be achieved by devising innovative pharmaceuticals or therapeutic approaches that can induce ferroptosis. The comprehensive utilization of the dual antitumor properties of ferroptosis offers a promising approach to enhance immunotherapy in the context of HCC.
Radiotherapy
Radioresistance in hepatocellular carcinoma is a leading cause of treatment failure. Regulating ferroptosis could reverse the radiation tolerance of HCC cells,101 improving the effectiveness of HCC radiotherapy. Radiotherapy elicited ferroptotic response in HCC cells through the downregulation of proteins that impede ferroptosis and the upregulation of proteins that facilitated it, consequently augmenting their susceptibility to radiation.102 Radiotherapy downregulated COMM domain containing 10 expressions, which inhibited its role in promoting hypoxia-inducible factor-1α (HIF1α) ubiquitination and degradation. This led to increased HIF1α expression and transcriptional promotion of SLC7A11, ultimately suppressing ferroptosis in HCC cells. The implantation of hydrogel scaffolds containing CuO nanoparticles, which were released in a controlled manner, activated ferroptosis after HCC resection. This activation occurred through Cu2+-induced depletion of glutathione and inactivation of GPX4, effectively preventing HCC recurrence after surgery.103 Changes in cellular copper levels also impacted the radiosensitivity of HCC cells. Radiotherapy can decrease the expression of COMM domain containing 10, which inhibits the degradation of HIF1α through ubiquitin. This leads to an increase in HIF1α expression and the suppression of ferroptosis in HCC cells, ultimately resulting in radioresistance. Additionally, the suppressor of cytokine signaling 2, an E3 ubiquitin ligase, can specifically induce the ubiquitination and degradation of the SLC7A11 protein, thereby promoting radiosensitization in HCC.104 Regulating ferroptosis-related proteins is crucial for enhancing the radiosensitivity of HCC, offering new perspectives for improving HCC radiotherapy effectiveness.
Conclusion
Ferroptosis has been discovered to be intricately associated with the progression and development of treatment resistance in HCC. HCC cells can resist therapeutic interventions by impeding the initiation of ferroptosis, a form of cell death induced by chemotherapeutic agents, targeted therapies, and radiotherapy, through multiple pathways. Conversely, these therapeutic modalities have the potential to directly or indirectly stimulate the activation of the ferroptosis pathway, thereby augmenting their cytotoxic effects or the effects of other medications. Hence, the manipulation of ferroptosis shows promise as a therapeutic approach to increase the susceptibility of HCC cells to different treatment methods. Further research can explore the connections between ferroptosis and the progression of HCC, as well as its role in drug resistance. This study may also give precedence to the exploration of novel pharmaceuticals with the ability to directly trigger ferroptosis, or investigate the potential synergistic impacts of combining these medications with established therapeutic approaches. By manipulating the ferroptosis process, these approaches aim to effectively address the difficulties associated with treating HCC and ultimately enhance patient survival rates.
Acknowledgments
This study was funded by the Jinhua traditional Chinese Medicine Science and Technology Research Program Project (2024LC11) and the Health Science and Technology Plan Project of Fujian Province (No. 2022QNA065). We express our gratitude for the provision of the picture materials by Figdraw (www.figdraw.com).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rumgay H, Arnold M, Ferlay J, et al. Global burden of primary liver cancer in 2020 and predictions to 2040. J Hepatol. 2022;77(6):1598–1606. doi:10.1016/j.jhep.2022.08.021
2. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. doi:10.1038/s41572-020-00240-3
3. Gordan JD, Kennedy EB, Abou-Alfa GK, et al. Systemic therapy for advanced hepatocellular carcinoma: ASCO guideline. J Clin Oncol. 2020;38(36):4317–4345. doi:10.1200/JCO.20.02672
4. Deng H, Kan A, Lyu N, et al. Tumor-derived lactate inhibit the efficacy of lenvatinib through regulating PD-L1 expression on neutrophil in hepatocellular carcinoma. J Immunother Cancer. 2021;9(6):e002305. doi:10.1136/jitc-2020-002305
5. Wang T, Qin Z-Y, Wen L-Z, et al. Epigenetic restriction of Hippo signaling by MORC2 underlies stemness of hepatocellular carcinoma cells. Cell Death Differ. 2018;25(12):2086–2100. doi:10.1038/s41418-018-0095-6
6. Di Giacomo S, Briz O, Monte MJ, et al. Chemosensitization of hepatocellular carcinoma cells to sorafenib by β-caryophyllene oxide-induced inhibition of ABC export pumps. Arch Toxicol. 2019;93(3):623–634. doi:10.1007/s00204-019-02395-9
7. Song Z, Liu T, Chen J, et al. HIF-1α-induced RIT1 promotes liver cancer growth and metastasis and its deficiency increases sensitivity to sorafenib. Cancer Lett. 2019;460:96–107. doi:10.1016/j.canlet.2019.06.016
8. Dong N, Shi X, Wang S, et al. M2 macrophages mediate sorafenib resistance by secreting HGF in a feed-forward manner in hepatocellular carcinoma. Br J Cancer. 2019;121(1):22–33. doi:10.1038/s41416-019-0482-x
9. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–125. doi:10.1038/s41422-020-00441-1
10. Shang Y, Luo M, Yao F, Wang S, Yuan Z, Yang Y. Ceruloplasmin suppresses ferroptosis by regulating iron homeostasis in hepatocellular carcinoma cells. Cell Signal. 2020;72:109633. doi:10.1016/j.cellsig.2020.109633
11. Zhu G, Murshed A, Li H, et al. O-GlcNAcylation enhances sensitivity to RSL3-induced ferroptosis via the YAP/TFRC pathway in liver cancer. Cell Death Discov. 2021;7(1):83. doi:10.1038/s41420-021-00468-2
12. Zhao Y, Li M, Yao X, et al. HCAR1/MCT1 regulates tumor ferroptosis through the lactate-mediated AMPK-SCD1 activity and its therapeutic implications. Cell Rep. 2020;33(10):108487. doi:10.1016/j.celrep.2020.108487
13. Wang Q, Guo Y, Wang W, et al. RNA binding protein DAZAP1 promotes HCC progression and regulates ferroptosis by interacting with SLC7A11 mRNA. Exp Cell Res. 2021;399(1):112453. doi:10.1016/j.yexcr.2020.112453
14. Yao F, Deng Y, Zhao Y, et al. A targetable LIFR-NF-ΰB-LCN2 axis controls liver tumorigenesis and vulnerability to ferroptosis. Nat Commun. 2021;12(1):7333. doi:10.1038/s41467-021-27452-9
15. Romualdo GR, Leroy K, Costa C, et al. In vivo and in vitro models of hepatocellular carcinoma: current strategies for translational modeling. Cancers. 2021;13(21):5583. doi:10.3390/cancers13215583
16. Sanchez-Martin A, Sanchon-Sanchez P, Romero MR, Marin JJG, Briz O. Impact of tumor suppressor genes inactivation on the multidrug resistance phenotype of hepatocellular carcinoma cells. Biomed Pharmacother. 2023;165:115209. doi:10.1016/j.biopha.2023.115209
17. Zhao S, Zhang Y, Lu X, et al. CDC20 regulates the cell proliferation and radiosensitivity of P53 mutant HCC cells through the Bcl-2/Bax pathway. Int J Biol Sci. 2021;17(13):3608–3621. doi:10.7150/ijbs.64003
18. Tang C, Qiu S, Mou W, Xu J, Wang P. Excessive activation of HOXB13/PIMREG axis promotes hepatocellular carcinoma progression and drug resistance. Biochem Biophys Res Commun. 2022;623:81–88. doi:10.1016/j.bbrc.2022.07.066
19. Marin JJG, Macias RIR, Monte MJ, et al. Molecular bases of drug resistance in hepatocellular carcinoma. Cancers. 2020;12(6):1663.
20. Buonanno M, De toledo SM, Pain D, Azzam EI. Long-term consequences of radiation-induced bystander effects depend on radiation quality and dose and correlate with oxidative stress. Radiat Res. 2011;175(4):405–415. doi:10.1667/RR2461.1
21. Wang J, Zhao H, Yu J, et al. MiR-320b/RAD21 axis affects hepatocellular carcinoma radiosensitivity to ionizing radiation treatment through DNA damage repair signaling. Cancer Sci. 2021;112(2):575–588. doi:10.1111/cas.14751
22. Liao J, Yi Y, Yue X, et al. Methyltransferase 1 is required for nonhomologous end-joining repair and renders hepatocellular carcinoma resistant to radiotherapy. Hepatology. 2023;77(6):1896–1910. doi:10.1002/hep.32615
23. Ladd AD, Duarte S, Sahin I, Zarrinpar A. Mechanisms of drug resistance in HCC. Hepatology. 2023;79(4):926–940. doi:10.1097/HEP.0000000000000237
24. Zheng T, Yin D, Lu Z, et al. Nutlin-3 overcomes arsenic trioxide resistance and tumor metastasis mediated by mutant p53 in Hepatocellular Carcinoma. Mol Cancer. 2014;13(1):133. doi:10.1186/1476-4598-13-133
25. Qian B, Che L, Du Z-B, et al. Protein phosphatase 2A-B55β mediated mitochondrial p-GPX4 dephosphorylation promoted sorafenib-induced ferroptosis in hepatocellular carcinoma via regulating p53 retrograde signaling. Theranostics. 2023;13(12):4288–4302. doi:10.7150/thno.82132
26. Lee JH, Suh JH, Kang HJ, et al. Tonicity-responsive enhancer-binding protein promotes stemness of liver cancer and cisplatin resistance. EBioMedicine. 2020;58:102926. doi:10.1016/j.ebiom.2020.102926
27. Chen C, Wang Z, Ding Y, Qin Y. Tumor microenvironment-mediated immune evasion in hepatocellular carcinoma. Front Immunol. 2023;14:1133308. doi:10.3389/fimmu.2023.1133308
28. Zhang S, Yuan L, Danilova L, et al. Spatial transcriptomics analysis of neoadjuvant cabozantinib and nivolumab in advanced hepatocellular carcinoma identifies independent mechanisms of resistance and recurrence. Genome Med. 2023;15(1):72. doi:10.1186/s13073-023-01218-y
29. Xing R, Gao J, Cui Q, Wang Q. Strategies to improve the antitumor effect of immunotherapy for hepatocellular carcinoma. Front Immunol. 2021;12:783236. doi:10.3389/fimmu.2021.783236
30. Zhou Z, Li X, Yang G, et al. Targeting β-catenin and PD-L1 simultaneously by a racemic supramolecular peptide for the potent immunotherapy of hepatocellular carcinoma. Theranostics. 2023;13(10):3371–3386. doi:10.7150/thno.83377
31. Chen S, Du Y, Guan X-Y, Yan Q. The current status of tumor microenvironment and cancer stem cells in sorafenib resistance of hepatocellular carcinoma. Front Oncol. 2023;13:1204513. doi:10.3389/fonc.2023.1204513
32. Wang J, Yu H, Dong W, et al. N6-methyladenosine-mediated up-regulation of FZD10 regulates liver cancer stem cells’ properties and lenvatinib resistance through WNT/β-catenin and hippo signaling pathways. Gastroenterology. 2023;164(6):990–1005.
33. Zhang D, Wu F, Song J, et al. A role for the NPM1/PTPN14/YAP axis in mediating hypoxia-induced chemoresistance to sorafenib in hepatocellular carcinoma. Cancer Cell Int. 2022;22(1):65. doi:10.1186/s12935-022-02479-0
34. Liao Y, Yang Y, Pan D, et al. HSP90α mediates sorafenib resistance in human hepatocellular carcinoma by necroptosis inhibition under hypoxia. Cancers. 2021;13(2):243. doi:10.3390/cancers13020243
35. Bamodu OA, Chang H-L, Ong J-R, Lee W-H, Yeh C-T, Tsai J-T. Elevated PDK1 expression drives PI3K/AKT/MTOR signaling promotes radiation-resistant and dedifferentiated phenotype of hepatocellular carcinoma. Cells. 2020;9(3):746. doi:10.3390/cells9030746
36. Raju B, Choudhary S, Narendra G, Verma H, Silakari O. Molecular modeling approaches to address drug-metabolizing enzymes (DMEs) mediated chemoresistance: a review. Drug Metab Rev. 2021;53(1):45–75. doi:10.1080/03602532.2021.1874406
37. Giannitrapani L, Di Gaudio F, Cervello M, et al. Genetic biomarkers of sorafenib response in patients with hepatocellular carcinoma. Int J Mol Sci. 2024;25(4):2197. doi:10.3390/ijms25042197
38. Caputo TM, Cusano AM, Principe S, et al. Sorafenib-loaded PLGA carriers for enhanced drug delivery and cellular uptake in liver cancer cells. Int J Nanomed. 2023;18:4121–4142. doi:10.2147/IJN.S415968
39. Wei J, Liu R, Zhang J, et al. Baicalin enhanced oral bioavailability of sorafenib in rats by inducing intestine absorption. Front Pharmacol. 2021;12:761763. doi:10.3389/fphar.2021.761763
40. Guo R, Lu F, Lin J, Fu C, Liu M, Yang S. Multi-b-value DWI to evaluate the synergistic antiproliferation and anti-heterogeneity effects of bufalin plus sorafenib in an orthotopic HCC model. Eur Radiol Exp. 2024;8(1):43. doi:10.1186/s41747-024-00448-y
41. Da X, Mo J, Li Q, et al. Targeted co-delivery of PD-L1 monoclonal antibody and sorafenib to circulating tumor cells via platelet-functionalized nanocarriers. Biochem Biophys Res Commun. 2023;671:335–342. doi:10.1016/j.bbrc.2023.05.124
42. Rigalli JP, Ciriaci N, Arias A, et al. Regulation of multidrug resistance proteins by genistein in a hepatocarcinoma cell line: impact on sorafenib cytotoxicity. PLoS One. 2015;10(3):e0119502. doi:10.1371/journal.pone.0119502
43. Yu G, Chen X, Chen S, Ye W, Hou K, Liang M. Arsenic trioxide reduces chemo-resistance to 5-fluorouracil and cisplatin in HBx-HepG2 cells via complex mechanisms. Cancer Cell Int. 2015;15(1):116. doi:10.1186/s12935-015-0269-y
44. Kong J, Qiu Y, Li Y, Zhang H, Wang W. TGF-β1 elevates P-gp and BCRP in hepatocellular carcinoma through HOTAIR/miR-145 axis. Biopharm Drug Dispos. 2019;40(2):70–80. doi:10.1002/bdd.2172
45. Lai S-C, Y-t S, Chi -C-C, et al. DNMT3b/OCT4 expression confers sorafenib resistance and poor prognosis of hepatocellular carcinoma through IL-6/STAT3 regulation. J Exp Clin Cancer Res. 2019;38(1):474. doi:10.1186/s13046-019-1442-2
46. Brandon EFA, Sparidans RW, Meijerman I, Manzanares I, Beijnen JH, Schellens JHM. In vitro characterization of the biotransformation of thiocoraline, a novel marine anti-cancer drug. Invest New Drugs. 2004;22(3):241–251. doi:10.1023/B:DRUG.0000026250.34645.7f
47. Kamdem LK, Flockhart DA, Desta Z. In vitro cytochrome P450-mediated metabolism of exemestane. Drug Metab Dispos. 2011;39(1):98–105. doi:10.1124/dmd.110.032276
48. Chen H, Shen Z-Y, Xu W, et al. Expression of P450 and nuclear receptors in normal and end-stage Chinese livers. World J Gastroenterol. 2014;20(26):8681–8690. doi:10.3748/wjg.v20.i26.8681
49. Guengerich FP. Cytochrome P450 enzymes as drug targets in human disease. Drug Metab Dispos. 2024;52(6):493–497. doi:10.1124/dmd.123.001431
50. Yang F, Hilakivi-Clarke L, Shaha A, et al. Metabolic reprogramming and its clinical implication for liver cancer. Hepatology. 2023;78(5):1602–1624. doi:10.1097/HEP.0000000000000005
51. Ishijima N, Kanki K, Shimizu H, Shiota G. Activation of AMP-activated protein kinase by retinoic acid sensitizes hepatocellular carcinoma cells to apoptosis induced by sorafenib. Cancer Sci. 2015;106(5):567–575. doi:10.1111/cas.12633
52. Fang Y, Zhan Y, Xie Y, et al. Integration of glucose and cardiolipin anabolism confers radiation resistance of HCC. Hepatology. 2022;75(6):1386–1401. doi:10.1002/hep.32177
53. Xu J, Ji L, Ruan Y, et al. UBQLN1 mediates sorafenib resistance through regulating mitochondrial biogenesis and ROS homeostasis by targeting PGC1β in hepatocellular carcinoma. Signal Transduct Target Ther. 2021;6(1):190. doi:10.1038/s41392-021-00594-4
54. Bergamini C, Leoni I, Rizzardi N, et al. MiR-494 induces metabolic changes through G6pc targeting and modulates sorafenib response in hepatocellular carcinoma. J Exp Clin Cancer Res. 2023;42(1):145. doi:10.1186/s13046-023-02718-w
55. Holmstrã¶m P, Gã¥fvels M, Eriksson LC, et al. Expression of iron regulatory genes in a rat model of hepatocellular carcinoma. Liver Int. 2006;26(8):976–985. doi:10.1111/j.1478-3231.2006.01316.x
56. Gao J, Chen J, Kramer M, Tsukamoto H, Zhang A-S, Enns CA. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. 2009;9(3):217–227. doi:10.1016/j.cmet.2009.01.010
57. Tseng -H-H, Chang J-G, Hwang Y-H, Yeh K-T, Chen Y-L, H-s Y. Expression of hepcidin and other iron-regulatory genes in human hepatocellular carcinoma and its clinical implications. J Cancer Res Clin Oncol. 2009;135(10):1413–1420. doi:10.1007/s00432-009-0585-5
58. Hao S-H, X-d M, Xu L, et al. Dual specific phosphatase 4 suppresses ferroptosis and enhances sorafenib resistance in hepatocellular carcinoma. Drug Resist Updat. 2024;73:101052. doi:10.1016/j.drup.2024.101052
59. Kim YW, Lee SM, Shin SM, et al. Efficacy of sauchinone as a novel AMPK-activating lignan for preventing iron-induced oxidative stress and liver injury. Free Radic Biol Med. 2009;47(7):1082–1092. doi:10.1016/j.freeradbiomed.2009.07.018
60. Saito Y, Yin D, Kubota N, et al. A therapeutically targetable TAZ-TEAD2 pathway drives the growth of hepatocellular carcinoma via ANLN and KIF23. Gastroenterology. 2023;164(7):1279–1292. doi:10.1053/j.gastro.2023.02.043
61. Qi R, Bai Y, Li K, et al. Cancer-associated fibroblasts suppress ferroptosis and induce gemcitabine resistance in pancreatic cancer cells by secreting exosome-derived ACSL4-targeting miRNAs. Drug Resist Updat. 2023;68:100960. doi:10.1016/j.drup.2023.100960
62. Yang Y, Gu H, Zhang K, et al. Exosomal ACADM sensitizes gemcitabine-resistance through modulating fatty acid metabolism and ferroptosis in pancreatic cancer. BMC Cancer. 2023;23(1):789. doi:10.1186/s12885-023-11239-w
63. Qu X, Liu B, Wang L, et al. Loss of cancer-associated fibroblast-derived exosomal DACT3-AS1 promotes malignant transformation and ferroptosis-mediated oxaliplatin resistance in gastric cancer. Drug Resist Updat. 2023;68:100936. doi:10.1016/j.drup.2023.100936
64. Kang X, Huo Y, Jia S, et al. Silenced LINC01134 enhances oxaliplatin sensitivity by facilitating ferroptosis through GPX4 in hepatocarcinoma. Front Oncol. 2022;12:939605. doi:10.3389/fonc.2022.939605
65. Zhan M, Ding Y, Huang S, et al. Lysyl oxidase-like 3 restrains mitochondrial ferroptosis to promote liver cancer chemoresistance by stabilizing dihydroorotate dehydrogenase. Nat Commun. 2023;14(1):3123. doi:10.1038/s41467-023-38753-6
66. Hu W, Zhou C, Jing Q, et al. FTH promotes the proliferation and renders the HCC cells specifically resist to ferroptosis by maintaining iron homeostasis. Cancer Cell Int. 2021;21(1):709. doi:10.1186/s12935-021-02420-x
67. Liu L, Lv Z, Wang M, Zhang D, Liu D, Zhu F. HBV enhances sorafenib resistance in hepatocellular carcinoma by reducing ferroptosis via SRSF2-mediated abnormal PCLAF splicing. Int J Mol Sci. 2023;24(4):3263.
68. Zhou Q-M, Y-f L, Zhou J-P, et al. Self-amplification of oxidative stress with tumour microenvironment-activatable iron-doped nanoplatform for targeting hepatocellular carcinoma synergistic cascade therapy and diagnosis. J Nanobiotechnology. 2021;19(1):361. doi:10.1186/s12951-021-01102-0
69. Corti A, Dominici S, Piaggi S, Pompella A. Enhancement of ferroptosis by boric acid and its potential use as chemosensitizer in anticancer chemotherapy. Biofactors. 2023;49(2):405–414. doi:10.1002/biof.1919
70. Wei W, Lu Y, Hu Q, et al. Synergistic antitumor efficacy of gemcitabine and cisplatin to induce ferroptosis in pancreatic ductal adenocarcinoma via Sp1-SAT1-polyamine metabolism pathway. Cell Oncol. 2023;47(1):321–341.
71. Zeng Y, Jiang H, Zhang X, et al. Canagliflozin reduces chemoresistance in hepatocellular carcinoma through PKM2-c-Myc complex-mediated glutamine starvation. Free Radic Biol Med. 2023;208:571–586. doi:10.1016/j.freeradbiomed.2023.09.006
72. Huang A, Yang X-R, Chung W-Y, Dennison AR, Zhou J. Targeted therapy for hepatocellular carcinoma. Signal Transduct Target Ther. 2020;5(1):146. doi:10.1038/s41392-020-00264-x
73. Yau T, Park J-W, Finn RS, et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): a randomised, multicentre, open-label, Phase 3 trial. Lancet Oncol. 2022;23(1):77–90. doi:10.1016/S1470-2045(21)00604-5
74. Zhu Y-J, Zheng B, Wang H-Y, Chen L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin. 2017;38(5):614–622. doi:10.1038/aps.2017.5
75. Liu M-R, Shi C, Song Q-Y, et al. Sorafenib induces ferroptosis by promoting TRIM54-mediated FSP1 ubiquitination and degradation in hepatocellular carcinoma. Hepatol Commun. 2023;7(10). doi:10.1097/HC9.0000000000000246
76. Sun J, Zhou C, Zhao Y, et al. Quiescin sulfhydryl oxidase 1 promotes sorafenib-induced ferroptosis in hepatocellular carcinoma by driving EGFR endosomal trafficking and inhibiting NRF2 activation. Redox Biol. 2021;41:101942. doi:10.1016/j.redox.2021.101942
77. Elkateb AS, Nofal S, Ali SA, Atya HB. Camptothecin sensitizes hepatocellular carcinoma cells to sorafenib- induced ferroptosis via suppression of Nrf2. Inflammation. 2023;46(4):1493–1511. doi:10.1007/s10753-023-01823-4
78. Iseda N, Itoh S, Yoshizumi T, et al. Impact of nuclear factor erythroid 2-related factor 2 in hepatocellular carcinoma: cancer metabolism and immune status. Hepatol Commun. 2022;6(4):665–678. doi:10.1002/hep4.1838
79. Sun X, Ou Z, Chen R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63(1):173–184. doi:10.1002/hep.28251
80. Sun X, Niu X, Chen R, et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016;64(2):488–500. doi:10.1002/hep.28574
81. Ding Z, Pan Y, Shang T, et al. URI alleviates tyrosine kinase inhibitors-induced ferroptosis by reprogramming lipid metabolism in p53 wild-type liver cancers. Nat Commun. 2023;14(1):6269. doi:10.1038/s41467-023-41852-z
82. Shi Z, Li Z, Jin B, et al. Loss of LncRNA DUXAP8 synergistically enhanced sorafenib induced ferroptosis in hepatocellular carcinoma via SLC7A11 de-palmitoylation. Clin Transl Med. 2023;13(6):e1300. doi:10.1002/ctm2.1300
83. Zhang L, X-m L, Shi X-H, et al. Sorafenib triggers ferroptosis via inhibition of HBXIP/SCD axis in hepatocellular carcinoma. Acta Pharmacol Sin. 2023;44(3):622–634. doi:10.1038/s41401-022-00981-9
84. Hu X, He Y, Han Z, et al. PNO1 inhibits autophagy-mediated ferroptosis by GSH metabolic reprogramming in hepatocellular carcinoma. Cell Death Dis. 2022;13(11):1010. doi:10.1038/s41419-022-05448-7
85. Xiao Y, Xu Z, Cheng Y, et al. Fe3+-binding transferrin nanovesicles encapsulating sorafenib induce ferroptosis in hepatocellular carcinoma. Biomater Res. 2023;27(1):63. doi:10.1186/s40824-023-00401-x
86. Ren X, Li Y, Zhou Y, et al. Overcoming the compensatory elevation of NRF2 renders hepatocellular carcinoma cells more vulnerable to disulfiram/copper-induced ferroptosis. Redox Biol. 2021;46:102122. doi:10.1016/j.redox.2021.102122
87. Wang Q, Bin C, Xue Q, et al. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021;12(5):426. doi:10.1038/s41419-021-03718-4
88. Lai Y, Lu N, Luo S, Wang H, Zhang P. A photoactivated sorafenib-Ruthenium(II) prodrug for resistant hepatocellular carcinoma therapy through ferroptosis and purine metabolism disruption. J Med Chem. 2022;65(19):13041–13051. doi:10.1021/acs.jmedchem.2c00880
89. Tang H, Chen D, Li C, et al. Dual GSH-exhausting sorafenib loaded manganese-silica nanodrugs for inducing the ferroptosis of hepatocellular carcinoma cells. Int J Pharm. 2019;572:118782. doi:10.1016/j.ijpharm.2019.118782
90. Kim D-H, Kim M-J, Kim N-Y, et al. DN200434, an orally available inverse agonist of estrogen-related receptor γ, induces ferroptosis in sorafenib-resistant hepatocellular carcinoma. BMB Rep. 2022;55(11):547–552. doi:10.5483/BMBRep.2022.55.11.089
91. Iseda N, Itoh S, Toshida K, et al. Ferroptosis is induced by lenvatinib through fibroblast growth factor receptor-4 inhibition in hepatocellular carcinoma. Cancer Sci. 2022;113(7):2272–2287. doi:10.1111/cas.15378
92. Song Z, Zhang Y, Luo W, et al. HAND2-AS1 promotes ferroptosis to reverse lenvatinib resistance in hepatocellular carcinoma by TLR4/NOX2/DUOX2 Axis. Curr Cancer Drug Targets. 2024;24. doi:10.2174/0115680096279597240219055135
93. Zhang N, Yang X, Piao M, et al. Biomarkers and prognostic factors of PD-1/PD-L1 inhibitor-based therapy in patients with advanced hepatocellular carcinoma. Biomark Res. 2024;12(1):26. doi:10.1186/s40364-023-00535-z
94. Itoh S, Yoshizumi T, Yugawa K, et al. Impact of immune response on outcomes in hepatocellular carcinoma: association with vascular formation. Hepatology. 2020;72(6):1987–1999. doi:10.1002/hep.31206
95. Zheng Y, Wang Y, Lu Z, et al. PGAM1 inhibition promotes HCC ferroptosis and synergizes with anti-PD-1 immunotherapy. Adv Sci. 2023;10(29):e2301928. doi:10.1002/advs.202301928
96. Qiu Y, Wu Z, Chen Y, et al. Nano ultrasound contrast agent for synergistic chemo-photothermal therapy and enhanced immunotherapy against liver cancer and metastasis. Adv Sci. 2023;10(21):e2300878. doi:10.1002/advs.202300878
97. Zhang D, Man D, Lu J, et al. Mitochondrial TSPO promotes hepatocellular carcinoma progression through ferroptosis inhibition and immune evasion. Adv Sci. 2023;10(15):e2206669.
98. Conche C, Finkelmeier F, Peå¡iä‡ M, et al. Combining ferroptosis induction with MDSC blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut. 2023;72(9):1774–1782. doi:10.1136/gutjnl-2022-327909
99. Tang B, Zhu J, Wang Y, et al. Targeted xCT-mediated ferroptosis and protumoral polarization of macrophages is effective against HCC and enhances the efficacy of the anti-PD-1/L1 response. Adv Sci. 2023;10(2):e2203973. doi:10.1002/advs.202203973
100. Hao X, Zheng Z, Liu H, et al. Inhibition of APOC1 promotes the transformation of M2 into M1 macrophages via the ferroptosis pathway and enhances anti-PD1 immunotherapy in hepatocellular carcinoma based on single-cell RNA sequencing. Redox Biol. 2022;56:102463. doi:10.1016/j.redox.2022.102463
101. Li Y, Yang W, Zheng Y, et al. Targeting fatty acid synthase modulates sensitivity of hepatocellular carcinoma to sorafenib via ferroptosis. J Exp Clin Cancer Res. 2023;42(1):6. doi:10.1186/s13046-022-02567-z
102. Yang M, Wu X, Hu J, et al. COMMD10 inhibits HIF1α/CP loop to enhance ferroptosis and radiosensitivity by disrupting Cu-Fe balance in hepatocellular carcinoma. J Hepatol. 2022;76(5):1138–1150. doi:10.1016/j.jhep.2022.01.009
103. Dang W, Chen W-C, Ju E, et al. 3D printed hydrogel scaffolds combining glutathione depletion-induced ferroptosis and photothermia-augmented chemodynamic therapy for efficiently inhibiting postoperative tumor recurrence. J Nanobiotechnology. 2022;20(1):266. doi:10.1186/s12951-022-01454-1
104. Chen Q, Zheng W, Guan J, et al. SOCS2-enhanced ubiquitination of SLC7A11 promotes ferroptosis and radiosensitization in hepatocellular carcinoma. Cell Death Differ. 2023;30(1):137–151. doi:10.1038/s41418-022-01051-7
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Identification of a Novel Ferroptosis-Related Gene Signature for Predicting Prognosis and Responsiveness to Immunotherapy in Hepatocellular Carcinoma
Wang Q, Wang B, Ma X, Zhuang H, Xie Z, Tang C, Tan W, Yang L, Shang C, Chen Y
Journal of Hepatocellular Carcinoma 2023, 10:1-16
Published Date: 10 January 2023
Identification and Validation of Ferroptosis-Related Subtypes and a Predictive Signature in Hepatocellular Carcinoma
Zheng C, Peng Y, Wang H, Wang Y, Liu L, Zhao Q
Pharmacogenomics and Personalized Medicine 2023, 16:39-58
Published Date: 26 January 2023
Significance of Physical Status and Liver Function Reserve for Outcome of Patients with Advanced Hepatocellular Carcinoma Receiving Lenvatinib Treatment
Chan KM, Lai Y, Hung HC, Lee JC, Cheng CH, Wang YC, Wu TH, Lee CF, Wu TJ, Chou HS, Wang CT, Chai PM, Lien HY, Lee WC
Journal of Hepatocellular Carcinoma 2023, 10:281-290
Published Date: 18 February 2023
Synergistic Effect of Lenvatinib and Chemotherapy in Hepatocellular Carcinoma Using Preclinical Models
Wang M, Yao X, Bo Z, Zheng J, Yu H, Xie X, Lin Z, Wang Y, Chen G, Wu L
Journal of Hepatocellular Carcinoma 2023, 10:483-495
Published Date: 27 March 2023
Breast Cancer: An Overview of Current Therapeutic Strategies, Challenge, and Perspectives
Wang J, Wu SG
Breast Cancer: Targets and Therapy 2023, 15:721-730
Published Date: 20 October 2023