")
Back to Journals » Cancer Management and Research » Volume 16
Deguelin Restores Paclitaxel Sensitivity in Paclitaxel-Resistant Ovarian Cancer Cells via Inhibition of the EGFR Signaling Pathway
Authors Bae S, Bae S, Kim HS, Lim YJ, Kim G, Park IC, So KA, Kim TJ, Lee JH
Received 29 December 2023
Accepted for publication 21 May 2024
Published 28 May 2024 Volume 2024:16 Pages 507—525
DOI https://doi.org/10.2147/CMAR.S457221
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Seunghee Bae,1 Sowon Bae,1 Hee Su Kim,1 Ye Jin Lim,1 Gyeongmi Kim,2 In-Chul Park,2 Kyeong A So,3 Tae Jin Kim,3 Jae Ho Lee1
1Department of Cosmetics Engineering, Konkuk University, Seoul, 05029, Republic of Korea; 2Division of Fusion Radiology Research, Korea Institute of Radiological & Medical Sciences, Seoul, 01812, Republic of Korea; 3Department of Obstetrics and Gynecology, Konkuk University School of Medicine, Seoul, 05030, Republic of Korea
Correspondence: Jae Ho Lee, Department of Cosmetics Engineering, Konkuk University, Seoul, 05029, Republic of Korea, Email [email protected]
Background: Ovarian cancer is one of women’s malignancies with the highest mortality among gynecological cancers. Paclitaxel is used in first-line ovarian cancer chemotherapy. Research on paclitaxel-resistant ovarian cancer holds significant clinical importance.
Methods: Cell viability and flow cytometric assays were conducted at different time and concentration points of deguelin and paclitaxel treatment. Immunoblotting was performed to assess the activation status of key signaling molecules important for cell survival and proliferation following treatment with deguelin and paclitaxel. The fluo-3 acetoxymethyl assay for P-glycoprotein transport activity assay and cell viability assay in the presence of N-acetyl-L-cysteine were also conducted.
Results: Cell viability and flow cytometric assays demonstrated that deguelin resensitized paclitaxel in a dose- and time-dependent manner. Cotreatment with deguelin and paclitaxel inhibited EGFR and its downstream signaling molecules, including AKT, ERK, STAT3, and p38 MAPK, in SKOV3-TR cells. Interestingly, cotreatment with deguelin and paclitaxel suppressed the expression level of EGFR via the lysosomal degradation pathway. Cotreatment did not affect the expression and function of P-glycoprotein. N-acetyl-L-cysteine failed to restore cell cytotoxicity when used in combination with deguelin and paclitaxel in SKOV3-TR cells. The expression of BCL-2, MCL-1, and the phosphorylation of the S155 residue of BAD were downregulated. Moreover, inhibition of paclitaxel resistance by deguelin was also observed in HeyA8-MDR cells.
Conclusion: Our research showed that deguelin effectively suppresses paclitaxel resistance in SKOV3-TR ovarian cancer cells by downregulating the EGFR and its downstream signaling pathway and modulating the BCL-2 family proteins. Furthermore, deguelin exhibits inhibitory effects on paclitaxel resistance in HeyA8-MDR ovarian cancer cells, suggesting a potential mechanism for paclitaxel resensitization that may not be cell-specific. These findings suggest that deguelin holds promise as an anticancer therapeutic agent for overcoming chemoresistance in ovarian cancer.
Keywords: ovarian cancer, paclitaxel-resistance, deguelin, EGFR, AKT
Graphical Abstract:
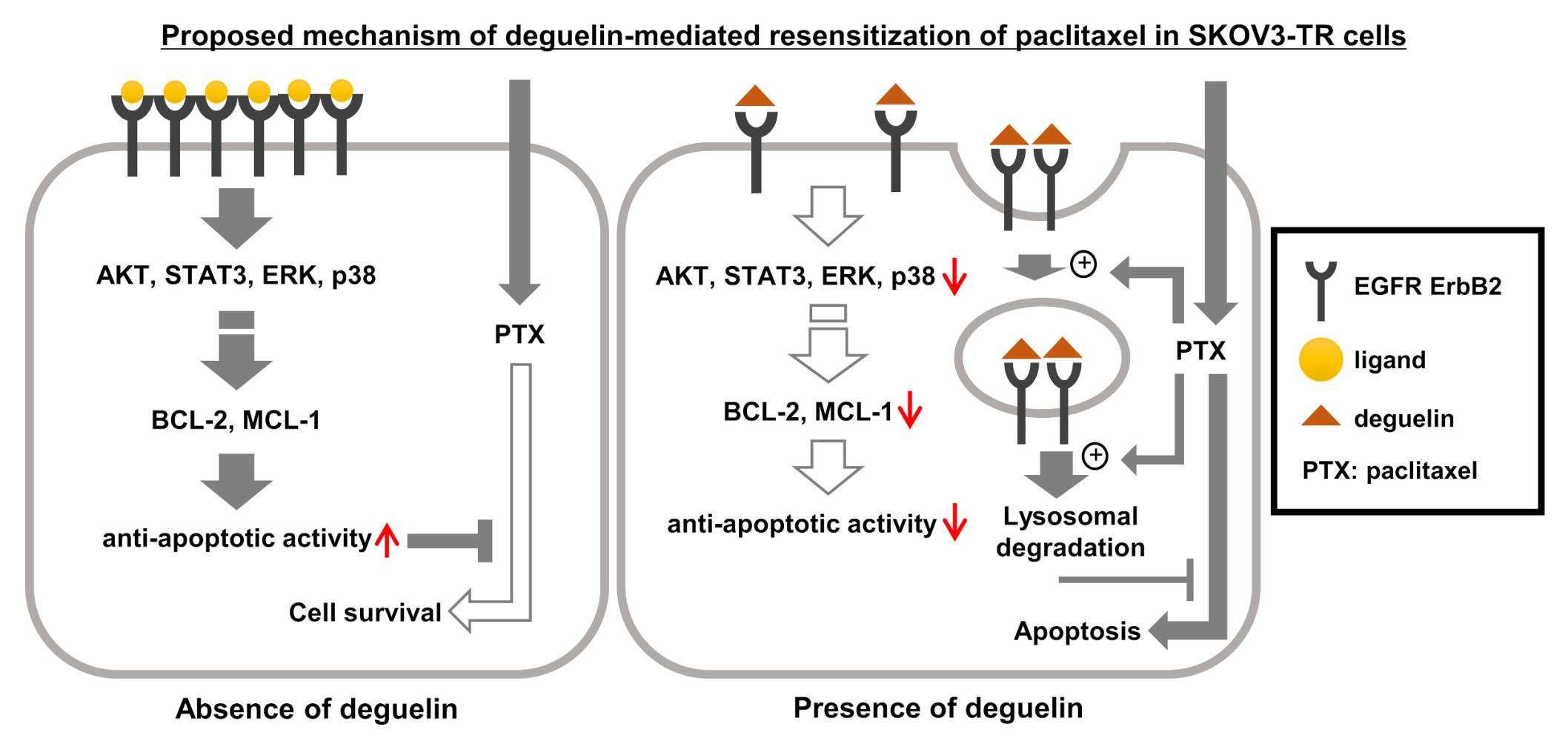
Introduction
Ovarian cancer is one of the most lethal gynecological diseases, with a five-year survival rate of only 30%.1 Developing biomarkers for early diagnosis and treatment of ovarian cancer is considered important to overcome ovarian cancer economically and cost-effectively.2 Epithelial ovarian cancer, which accounts for approximately 90% of malignant ovarian cancer, presents as a heterogeneous gynecological disease that has been classified into six histotypes, including serous, mucinous, endometrioid, clear cell, seromucinous, and transitional cells (Brenner).3,4 Histopathological and molecular genetic studies have provided a subdivision of epithelial ovarian cancer into two groups: Type I ovarian carcinoma is defined as a relatively slow-growing and indolent cancer with a precursor lesion in the ovary, and type II carcinoma is a more aggressive and genetically unstable cancer that develops de novo from serous tubal intraepithelial carcinoma or ovarian epithelium.5 Type I carcinomas include low-grade serous, mucinous, endometrioid, clear cell, and Brenner carcinomas, while type II carcinomas include high-grade serous carcinoma, undifferentiated, and carcinosarcoma.6 Type I and type II ovarian carcinomas have different molecular genetic characterizations. High-grade serous ovarian carcinoma is the most common type of ovarian carcinoma (type II carcinoma), which is frequently mutated in TP53 and BRCA, but not in genes that are predominantly mutated in type I carcinomas such as KRAA, BRAF, ERBB2, PTEN, CTNNB1, and PIK3CA.7 Paclitaxel has been employed as the initial chemotherapeutic option for these specific ovarian cancer types.8 Nonetheless, the emergence of chemoresistance remains a significant challenge to the effectiveness of ovarian cancer chemotherapy.9 While CA125 and HE4 measurements are currently approved diagnostic tools for ovarian cancer, they are insufficient for early detection.10 The discovery of suitable biomarkers for the development of new therapeutic agents and the diagnosis of chemosensitivity to paclitaxel to overcome chemoresistance have been considered important research areas aiming to improve ovarian cancer patient outcomes.
Paclitaxel, a natural product originally derived from Taxus brevifolia,11 has been extensively used as an anticancer drug against several malignant cancers, including advanced ovarian and triple-negative breast carcinomas.12 Previous studies have demonstrated that paclitaxel induces apoptotic cell death through different mechanisms or apoptotic pathways in multiple cell types.13,14 Paclitaxel stabilizes cellular microtubules and inhibits the formation of the mitotic spindle.15 Consequently, p53 is induced in cells and eventually activates an apoptotic pathway called mitotic catastrophe.16 In addition, it has been reported that there are various pathways by which paclitaxel can induce apoptosis through p53-independent mechanisms.17 Paclitaxel induces apoptosis by accumulating reactive oxygen species (ROS) and downregulating the ROS-scavenging enzyme superoxide dismutase (SOD) in canine mammary gland tumor cells.18 Also, paclitaxel induces apoptosis in breast cancer cells through the calcium efflux mechanism.19 Miller et al reported that paclitaxel-induced cell death requires the intrinsic apoptotic pathway, including BAK and MCL-1, in breast cancer cells.20 Several reports have demonstrated that PI3K/AKT, MAPK, and STAT3 signaling, which are key cellular mediators for cell survival from various stress, are significantly involved in paclitaxel-induced apoptosis in various cells.21,22
Epidermal growth factor receptors (EGFRs), members of the ErbB receptor family, consist of four members with a common structural backbone: EGFR (ErbB1, HER1), ErbB2 (HER2), ErbB3 (HER3), and ErbB4 (HER4).23 EGFR contains a cytoplasmic tyrosine-kinase domain, a transmembrane domain, and an extracellular ligand-binding domain.24 EGFR is able to form a variety of homo- or heterodimers, each combination modulating intracellular signaling while conferring diversity to different ligand affinities and receptor tyrosine kinase activity.25 EGFR signaling plays critical roles in various cellular events, including proliferation, differentiation, development, and apoptosis.26 The oncogenic property of EGFR was first identified when the v-ErbB viral oncogene of the avian erythroblastosis virus was found to be a homolog of the human EGFR gene.27 Many reports have indicated that EGFR is commonly upregulated in various cancers, including non-small-cell lung cancer, pancreatic cancer, glioblastoma, breast cancer, and head and neck cancer.28–33 Amplifications of the EGFR gene have been reported in more than half of glioblastoma multiforme (GBM), and it has been known that high levels of EGFR activity directly affect the malignancy of GBM.34 In non-small cell lung cancer (NSCLC), EGFR overexpression or intracellular EGFR mutations represent about 43–89% of cases.29 Also, it has been reported that 25% of NSCLC have mutations in the EGFR tyrosine kinase domain, correlating with increasing receptor expression in 75% of cases.35 Epithelial ovarian cancer has also been linked to amplifications and overexpression of multiple EGFR family members.36 Although anti-EGFR-targeted therapy has shown limited clinical activity in ovarian cancer to date,37 several reports have suggested that the expression level of EGFR is a prognostic biomarker for ovarian cancer.38,39 However, the effect of EGFR signaling on paclitaxel resistance in ovarian cancer has not been reported yet.
EGFR can modulate its activity level through spatiotemporal control of the receptor induced by endocytic trafficking.40 The regulation of EGFR expression occurs not only by the ubiquitination/proteasomal degradation pathway but also by the endocytosis/lysosomal degradation pathway.41–43 The strength of ligand-binding between the EGFR heterodimers could affect ligand dissociation efficiency in the endosome, which leads to lysosomal degradation of EGFR, affecting the stability of the EGFR family.41 In canonical ligand-dependent EGFR signaling, receptor endocytosis and recycling have been reported to be important for the temporal regulation of EGFR signaling.44 Also, clathrin-dependent or non-clathrin endocytic pathways are utilized differently depending on the ligand concentration or cellular environment to maintain or weaken the EGFR signaling mechanism.45 A number of studies suggest that the mechanisms of EGFR endocytosis and degradation, facilitated by small molecules, are stimulus-dependent and complex.46,47 Consequently, the exploration of mechanisms of EGFR degradation and the discovery of novel EGFR degraders are crucial for the development of new strategies to control EGFR-positive tumors.
Deguelin, a member of the rotenone family, is a compound isolated from Derris trifoliata Lour. or Mundulea sericea (Leguminosae).48 Rotenone, a highly toxic deguelin-analogue, has traditionally served as an insecticide and a herbicide for an extended period of time,49 and has been studied as an inducer of Parkinson’s disease.50,51 Despite their structural similarities to rotenone, several studies have reported that deguelin exhibits distinct aspects of toxicity and molecular function compared to rotenone.52–56 Accumulating evidence has supported the significant anticancer potential of the natural product deguelin across various human cancer models, including lung cancer,57 hepatocellular carcinoma,58 colorectal cancer,59 esophageal carcinoma,60 metastatic melanoma,61 and breast cancer.62 Molecular mechanisms underlying deguelin-mediated anti-tumor effects have been elucidated, involving the inhibition of Aurora B kinase to delay cell cycle progression, modulation of metabolic pathways such as glycolysis suppression, attenuation of angiogenesis and metastasis, and activation of intrinsic apoptosis.63–65 However, the effect of deguelin on paclitaxel-resistant ovarian cancer cells was not clear. In the present study, we first investigated the inhibitory effect of deguelin on paclitaxel resistance in SKOV3-TR cells.
Materials and Methods
Cell Culture and Reagents
Ovarian cancer cell lines in this study (SKOV3, SKOV3-TR, and HeyA8-MDR) were provided by Dr. Anil K. Sood (University of Texas MD Anderson Cancer Center, TX, USA). SKOV3 cells were subcultured in RPMI 1640 medium (Biowest, Nuaillé, France) supplemented with 10% fetal bovine serum (FBS) (Corning, NY, USA) and 1% penicillin/streptomycin (Gibco; Thermo Fisher Scientific, Waltham, MA, USA). SKOV3-TR and HeyA8-MDR cells were grown in culture medium with 50 nM paclitaxel (Cayman Chemical Company, Ann Arbor, MI, USA) to sustain their paclitaxel resistance. All cells were cultured in a humidified incubator at 37 °C with 5% CO2. Antibodies against AKT (#9272), phospho-AKT (Ser473) (#9271), ERK (#9102), phospho-ERK (Thr202/Tyr204) (#9101), p38 (#9212), phospho-p38 (Thr180/Tyr182) (#9211), STAT3 (#9139), phospho-STAT3 (Tyr705) (#9145), phospho-STAT3 (Ser727) (#94994), EGFR (#2232), phospho-EGFR (Tyr1068) (#2234), ERBB2 (#2165), phospho-ERBB2 (Tyr1248) (#2247), BCL-XL (#2764), cleaved caspase 3 (#9664), cleaved PARP (#5625), MCL-1 (#5453), BAX (#5023), survivin (#2803), phospho-BAD (Ser115) (#9297), and MDR1/ABCB1 (#12693) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Anti-BAD (sc-8044) and anti-Actin (sc-47778) antibodies were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Anti-BCL-2 antibody was obtained from ABclonal (Woburn, MA, USA). Anti-procaspase 3 antibody was from Abcam (Waltham, Boston, USA). Deguelin and paclitaxel were obtained from Cayman Chemical Company (MI, USA). Dimethyl sulfoxide (DMSO), N-acetyl-L-cysteine (NAC), Bafilomycin A1, MG132, and epidermal growth factor (EGF) were purchased from Sigma-Aldrich (Darmstadt, Germany). Gefitinib and erlotinib were from MedChemExpress (NJ, USA).
Cell Viability Assay Using Water-Soluble Tetrazolium (WST-1) Analysis and Crystal Violet Staining
WST-1 analysis was performed using EZ-Cytox (DoGenBio, Seoul, Republic of Korea) according to the manufacturer’s protocol. SKOV3-TR (5 x 103) and HeyA8-MDR (7 x 103) cells were seeded on 96-well plates and further cultured for 24 h. Cells were treated with serially diluted paclitaxel (0–1000 nM) together with deguelin (0, 5, and 10 μM), and further incubated for 48 h. The EZ-Cytox solution was diluted to 10% in the medium and subsequently added to each well. Cell viability was investigated by measuring the absorbance at 450 nm using a Synergy™ HTX Multi-Mode Microplate Reader (BioTek, Winooski, VT, USA). SKOV3-TR (4 x 104) and HeyA8-MDR (5 x 104) cells were seeded in 24-well plates and further incubated for 24 h. Cells were treated with serially diluted paclitaxel (0, 25, 50, 100, 200, and 400 nM) together with deguelin (5 and 10 µM). After 48 h of incubation, the cells were added to 500 μL of 0.2% crystal violet solution (BioPure, Seoul, Republic of Korea) in each well and stained for 30 min.
Flow Cytometry for Apoptosis Analysis
Apoptosis induced by combined treatment with deguelin and paclitaxel was analyzed by the Annexin V-FITC/PI apoptosis staining/detection kit (Abcam, Cambridge, UK) following the manufacturer’s protocols. SKOV3-TR (2 x 105) cells were treated with paclitaxel (200 nM), deguelin (10 µM), and cotreated with paclitaxel and deguelin for 48 h. Cells were resuspended in 1X binding buffer and incubated with Annexin V-FITC and PI staining solution for 5 min in the dark. The quantity of apoptotic cells was analyzed using BD FACSCalibur (BD Bioscience, Mountain View, CA, USA).
Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNAs were extracted using a Ribo-EX reagent (GeneAll Biotechnology Co., Ltd., Seoul, Republic of Korea), and cDNAs were synthesized from 1 µg of the total RNAs using oligo dT, 1.5 mM dNTP, 0.1 M DTT, 5X First Strand Buffer, and M-MLV reverse transcriptase (Thermo Fisher Scientific, Waltham, MA, USA). The EvaGreen qPCR Mix Plus (ROX) master mix from Solis BioDyne (Tartu, Estonia) was utilized in the RT-qPCR. The StepOnePlus™ Real-Time PCR System (Applied Biosystems, Waltham, MA, USA) was employed for the analysis of relative quantifications of mRNA for the target gene. The expression levels of mRNA were normalized to the expression level of glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) housekeeping gene, an internally expressed reference gene. The primers used for the amplification of specific genes in this study were listed in Supplementary Table 1.
Fluo-3/AM Assay (P-Glycoprotein Transport Assay)
SKOV3 (6 x 105) cells and SKOV3-TR (6 x 105) cells were incubated in culture medium for 24 h. Then, cells were treated with paclitaxel (200 nM), deguelin (10 µM), and a combination of deguelin (10 µM) and paclitaxel (200 nM), respectively. After treatment, cells were further incubated for 24 h. Following this, 4 µM fluo-3 acetoxymethyl (AM) solution (Sigma Aldrich, St. Louis, USA) was added, and the cells were further incubated for 1 h at 37 °C. The cells were visualized by UV microscopy (Zeiss, Oberkochen, Germany).
Statistical Analysis
All statistical analyses were performed in triplicate. All data are presented as the mean ± standard deviation (SD). Normally distributed data were evaluated using a one-way analysis of variance (ANOVA), as presented in the figure legends. For all results, values of *p < 0.05 and ***p < 0.001 were considered statistically significant.
Results
Deguelin Resensitized Paclitaxel in SKOV3-TR Ovarian Cancer Cells
We first validated the differential sensitivity of paclitaxel in SKOV3 and SKOV3-TR ovarian cancer cells. Cells were treated with serially diluted paclitaxel (0–1000 nM) for 48 h, and cell viability was examined using the WST-1 analysis (Figure 1A). As a result, SKOV3-TR cells showed cell viability of up to 80% or more even when treated with 1000 nM paclitaxel, while SKOV3 cells showed cell viability of less than 50% even at a concentration of 7.8125 nM paclitaxel. This indicates that SKOV3-TR cells are highly resistant to paclitaxel (Figure 1A). Next, we examined whether cell viability was affected by deguelin treatment alone in SKOV3-TR cells using the WST-1 analysis. SKOV3-TR cells were treated with deguelin at concentrations of 2.5, 5, 10, and 20 μM for 48 h. As shown in Figure 1B, the WST-1 analysis showed no significant effect on cell viability by deguelin at concentrations of up to 10 μM (p > 0.1), but 20% cytotoxicity was observed when treated with 20 μM deguelin. We examined the effect of deguelin on paclitaxel resistance in SKOV3-TR cells. Cells were treated with serially diluted paclitaxel (0–500 nM) together with dose-different combinations (0, 5, and 10 µM) of deguelin for 48 h. WST-1 analysis showed that deguelin effectively decreased cell viability in paclitaxel-treated SKOV3-TR cells in a dose-dependent manner (***p < 0.001) (Figure 1C). To determine whether the mechanism of deguelin-induced resensitization of paclitaxel was associated with apoptosis in SKOV3-TR cells, immunoblotting was performed to detect cleaved PARP. As shown in Figure 1D and E, the cleaved PARP proteins were effectively increased by deguelin in paclitaxel-treated SKOV3-TR cells in a time- and concentration-dependent manner, indicating that deguelin could restore the paclitaxel sensitivity in SKOV3-TR cells through the apoptotic pathway. Next, the effect of deguelin on restoring paclitaxel resistance in SKOV3-TR cells was confirmed by microscopic observation and FACS analysis. SKOV3-TR cells were treated with paclitaxel, deguelin, and the combination of paclitaxel and deguelin, respectively, for 48 h. Microscopic observations also showed that deguelin clearly resensitized the paclitaxel in SKOV3-TR cells (Figure 1F). As shown in Figure 1G, the level of apoptotic activity was analyzed by Annexin V-FITC and PI staining. The percentage of apoptotic cells was significantly increased in SKOV3-TR cells cotreated with paclitaxel and deguelin compared to SKOV3-TR cells treated with deguelin and paclitaxel alone (***p < 0.001). These results demonstrated that deguelin effectively restored paclitaxel sensitivity in SKOV3-TR ovarian cancer cells.
 |
Figure 1 Cotreatment with deguelin and paclitaxel induced cell cytotoxicity in SKOV3-TR ovarian cancer cells. (A) SKOV3 (5 x 103) and SKOV3-TR (5 x 103) cells were seeded in 96-well plates and further incubated for 24 h. Cells were treated with serially diluted paclitaxel (0–1000 nM) for 48 h. (B) SKOV3-TR cells were treated with deguelin (0, 2.5, 5, 10, and 20 µM) for 48 h. (C) SKOV3-TR (5 x 103) cells were seeded in a 96-well plate for 24 h, then treated with serially diluted paclitaxel (0–500 nM) together with dose-different combinations (0, 5, and 10 µM) of deguelin. The cell viability was examined by WST-1 analysis. (D) SKOV3-TR (5 x 105) cells were seeded in 100 mm plates. After 24 h, cells were treated with DMSO (mock), paclitaxel (200 nM), deguelin (10 µM), and cotreated with deguelin (10 µM) and paclitaxel (200 nM) for 12 h, 24 h, and 48 h, respectively. (E) SKOV3-TR cells were treated with paclitaxel (200 nM) together with dose-different combinations (0, 1, 5, and 10 µM) of deguelin, and treated with dose-different combinations (0, 10, 100, and 200 nM) of paclitaxel together with deguelin (10 µM). At 48 h after treatment, cells were subjected to immunoblotting to examine the level of cleaved PARP. Actin was used as a loading control. (F) SKOV3-TR cells were treated with DMSO (mock), paclitaxel (200 nM), deguelin (10 µM), and treated with a combination of paclitaxel (200 nM) and deguelin (10 µM) for 48 h. The morphological changes of SKOV3-TR cells were observed by microscopy at 48 h. (G) SKOV3-TR (2 x 105) cells were seeded in 6-well plates and treated with paclitaxel (200 nM) and deguelin (10 µM) for 48 h. Living, early apoptotic, and late apoptotic cells were determined using Annexin V-FITC and PI staining, followed by flow cytometry. The statistical experiments were repeated three times. *p < 0.05 and ***p < 0.001 analyzed by concentration were considered significant. |
 |
Figure 2 Inhibition of phosphorylation of AKT, STAT3, ERK, and p38 MAPK by cotreatment of deguelin and paclitaxel in SKOV3-TR ovarian cancer cells. SKOV3-TR cells were treated with DMSO (mock), paclitaxel (200 nM), deguelin (10 µM), and cotreated with paclitaxel (200 nM) and deguelin (10 µM), respectively. At 48 h after treatment, cells were harvested, and then the expression of AKT, STAT3, p38 MAPK, and p42/44 ERK and their phosphorylation status were examined by immunoblotting. Actin was used as a loading control. |
 |
Figure 3 Downregulation of EGFR by cotreatment of deguelin and paclitaxel via the lysosomal degradation pathway in SKOV3-TR cells. (A) SKOV3-TR cells were treated with DMSO (mock), paclitaxel (200 nM), deguelin (10 µM), and cotreated with paclitaxel (200 nM) and deguelin (10 µM) as indicated for 48 h. The expression and phosphorylation levels of the EGFR family, including EGFR and ERBB2, were examined by immunoblotting. Actin was used as a loading control. (B) The transcription levels of EGFR and ERBB2 were examined by RT-qPCR in the same treatment. The mRNA expression of GAPDH was used as a loading control. (C) SKOV3-TR cells were treated with paclitaxel (200 nM) together with dose-different combinations (0, 1, 5, and 10 µM) of deguelin and treated with dose-different combinations (0, 10, 100, and 200 nM) of paclitaxel together with deguelin (10 µM) for 48 h. And then the expression levels of EGFR and ERBB2 were examined by immunoblotting. (D) SKOV3-TR cells were cotreated with paclitaxel and deguelin. After 24 h of treatment, cells were additionally treated with DMSO, the lysosomal degradation inhibitor bafilomycin A1, and the proteasome inhibitor MG132 for 24 h. The expression levels of EGFR and ERBB2 were analyzed by immunoblotting. The statistical experiments were repeated three times. |
 |
Figure 4 The expression of P-glycoprotein (P-gp) and its efflux function were not affected by deguelin or paclitaxel in SKOV3-TR cells. (A) The expression levels of P-gp in SKOV3 and SKOV3-TR cells were confirmed by immunoblotting. Actin was used as a loading control. (B) SKOV3-TR cells were treated with paclitaxel, deguelin, and cotreated with paclitaxel and deguelin as indicated. At 48 h posttreatment, the expressions of P-gp were examined by immunoblotting. (C) SKOV3-TR cells were treated with paclitaxel and deguelin as above. At 48 h of treatment, cells were additionally treated with the fluorogenic P-gp substrate fluo-3/AM for 1 h. Fluorescent SKOV3-TR cells were visualized by UV microscopy. SKOV3 cells were also treated with fluo-3/AM as a positive control. |
 |
Figure 5 The expression levels of BCL-2 and MCL-1 were downregulated by cotreatment with deguelin and paclitaxel in SKOV3-TR cells. SKOV3-TR cells were seeded for 24 h and then treated with serially diluted paclitaxel (0–500 nM) together with deguelin (0 and 10 µM). At 24 h of treatment, cells were additionally treated with 5 mM NAC for 24 h. The cell viability was examined by WST-1 analysis (A) and a crystal violet assay (B). (C) SKOV3-TR cells were treated with paclitaxel, deguelin, and cotreated with deguelin and paclitaxel as indicated. At 48 h of treatment, the expression levels of BCL-2 family proteins, including BCL-2, BCL-XL, MCL-1, BAX, BAD, phospho-BAD (S155), and survivin, were examined by immunoblotting. Also, procaspase 3, cleaved caspase 3, and cleaved PARP were analyzed as apoptosis indicators. Actin was used as a loading control. (D) The transcriptions of BCL-2 family genes were examined by RT-qPCR. The mRNA expression of GAPDH was used as a loading control. The statistical experiments were repeated three times. *p < 0.05 and ***p < 0.001 analyzed by concentration were considered significant. |
 |
Figure 6 Cell cytotoxicity was induced by cotreatment of deguelin and paclitaxel in paclitaxel-resistant ovarian cancer HeyA8-MDR cells. (A) HeyA8-MDR (2 x 104) cells were seeded in a 96-well plate for 24 h, then treated with serially diluted paclitaxel (0–500 nM) together with dose-different combinations (0, 5, and 10 µM) of deguelin. The cell viability was examined by WST-1 analysis. (B) HeyA8-MDR (1 x 105) cells were seeded in a 24-well plate for 24 h, then treated with paclitaxel (0, 25, 100, 200, and 400 nM) together with dose-different combinations (0, 5, and 10 µM) of deguelin for 48 h. Then, cells were fixed and stained with a crystal violet solution. (C) HeyA8-MDR cells were treated with DMSO (mock), paclitaxel (200 nM), deguelin (10 µM), and cotreated with deguelin (10 µM) and paclitaxel (200 nM) as indicated for 48 h. The expression levels of EGFR were examined by immunoblotting. Actin was used as a loading control. The statistical experiments were repeated three times. ***p < 0.001 analyzed by concentration was considered significant. |
Deguelin Inhibited the Phosphorylation of AKT, STAT3, ERK, and p38 MAPK in Paclitaxel-Treated SKOV3-TR Cells
According to previous reports, the pivotal role of intracellular signaling molecules in affecting cell proliferation and survival has been identified, and their expression and phosphorylation levels are known to play an important role in the chemoresistance of various cancer cells.22,66–68 Therefore, we investigated the expression and phosphorylation levels of cellular signaling kinases involved in cell proliferation and survival, including AKT, STAT3, ERK, and p38 MAPK, through treatment with deguelin and paclitaxel in SKOV3-TR cells. Immunoblotting showed that the expression and phosphorylation of these molecules were not affected in SKOV3-TR cells treated with either paclitaxel or deguelin alone compared to control (Figure 2). However, the phosphorylation levels of AKT, STAT3, ERK, and p38 MAPK were inhibited in SKOV3-TR cells cotreated with deguelin and paclitaxel (Figure 2). These data revealed that deguelin can simultaneously inhibit AKT, STAT3, ERK, and p38 MAPK, crucial for cell proliferation and survival, and may also affect upstream signaling mechanisms common to their activation in paclitaxel-treated SKOV3-TR cells.
Deguelin Inhibited the Expression Level of EGFR in Paclitaxel-Treated SKOV3-TR Ovarian Cancer Cells via Enhancement of the Lysosomal Degradation Pathway
Recently, several studies have reported that deguelin can inhibit EGFR signaling and induce apoptosis in malignant cancer cells, including non-small cell lung cancer,69 breast cancer,70 and head and neck squamous cell carcinomas.71 Although apoptosis of SKOV3-TR ovarian cancer cells was not observed by treatment with deguelin alone in the above experimental results, we tested whether the resensitization of paclitaxel by deguelin was due to inhibition of EGFR signaling in SKOV3-TR cells. Immunoblotting showed that the expression of EGFR protein and its phosphorylation at the Tyr1068 residue were decreased by deguelin in paclitaxel-treated SKOV3-TR cells (Figure 3A). However, no significant differences in transcriptional expression were observed by RT-qPCR (Figure 3B). In addition, the expression and its phosphorylation at the Tyr1248 residue of ERBB2, one of the EGFR families, were also inhibited in SKOV3-TR cells cotreated with deguelin and paclitaxel in the same pattern of EGFR (Figure 3A and B). Also, we confirmed that the expressions of EGFR and ERBB2 in SKOV3-TR cells by cotreatment with deguelin and paclitaxel were inhibited in a concentration-dependent manner by deguelin and paclitaxel, respectively (Figure 3C). We next analyzed whether the decrease in EGFR expression by the cotreatment of deguelin and paclitaxel was caused by the ubiquitination/proteasomal degradation pathway or by the lysosomal degradation pathway. As shown in Figure 3D, immunoblotting showed that treatment with bafilomycin A1, which inhibits endosome acidification by inhibiting vacuolar H+-ATPase and thus prevents lysosomal degradation, restored the expression of EGFR in deguelin and paclitaxel-cotreated SKOV3-TR cells. In contrast, MG132, a proteasome inhibitor, did not affect the status of the expression of EGFR in deguelin and paclitaxel-treated SKOV3-TR cells (Figure 3D). Collectively, these results indicated that deguelin effectively inhibited the expression of EGFR by downregulating protein stability via the lysosomal degradation pathway in paclitaxel-treated SKOV3-TR cells.
Deguelin Did Not Affect the Expression and Function of P-Glycoprotein in SKOV3-TR Cells
It has been known that the most common mechanism of multidrug resistance in cancer is the overexpression of drug efflux transporters of the ATP binding cassette (ABC) family.72 P-glycoprotein (P-gp/MDR1) is a 170 kDa transmembrane protein and one of the members of the ABC family.73 P-gp-mediated drug efflux has been widely recognized as a major driver of resistance to anticancer agents including paclitaxel, cisplatin, and doxorubicin.74,75 We investigated whether deguelin may reduce paclitaxel resistance in SKOV3-TR cells by modulating the expression of P-gp or its function. Immunoblotting showed that P-gp was overexpressed in SKOV3-TR cells (Figure 4A), but the cotreatment of deguelin and paclitaxel did not affect the expression of P-gp in SKOV3-TR cells (Figure 4B). The cell-permeant dye fluo-3/AM is a fluorescent substrate for P-gp, and several reports have used it to measure the transport activity of P-gp.76,77 SKOV3-TR cells were treated with deguelin and paclitaxel alone or cotreated with deguelin and paclitaxel, and then further incubated with 4 μM fluo-3/AM for an additional 1 h. SKOV3-TR cells did not internalize the fluorescent dye compared to SKOV3 cells, as indicated by UV-microscopy, showing that fluo-3/AM was effectively excluded in SKOV3-TR cells (Figure 4C). However, the uptake of fluo-3/AM fluorescence was not increased either alone or cotreated with deguelin and paclitaxel in SKOV3-TR cells, indicating that deguelin did not affect the function of P-gp in SKOV3-TR cells (Figure 4C). These results revealed that cotreatment of deguelin with paclitaxel did not inhibit the expression of P-gp or substrate transport function in SKOV3-TR cells.
Deguelin Suppressed the Expression of BCL-2 and MCL-1 and the Phosphorylation of BAD in SKOV3-TR Cells
Next, we attempted to examine the mechanism by which deguelin inhibits paclitaxel resistance in SKOV3-TR cells. Several reports indicated that the mechanism of cell death caused by paclitaxel treatment may be due to excessive induction of intracellular ROS.18,78 To determine whether ROS mediates the cell cytotoxicity in SKOV3-TR cells cotreated with deguelin and paclitaxel, we additionally treated NAC and then examined cell viability by WST-1 analysis. Figure 5A showed that NAC did not inhibit cell cytotoxicity induced by cotreatment with deguelin and paclitaxel in SKOV3-TR cells. Crystal violet staining also indicated that NAC did not restore cell viability in SKOV3-TR cells cotreated with deguelin and paclitaxel (Figure 5B). It has been reported that upon ligand recognition, EGFR directly recruits a class I phosphatidylinositol 3-kinase (PI3K), followed by activation of AKT.22 Moreover, AKT has been reported as a significant signaling effector for several pathways, including cell growth, modulation of metabolism, and inhibition of apoptosis.79,80 Since it has been reported that the PI3K/AKT pathway is important for multidrug resistance processes in various cancer cells via the inhibition of apoptosis,81 we next examined whether deguelin may modulate BCL-2 family proteins. Immunoblotting indicated that the expression of BCL-2, MCL-1, and the phosphorylation of BAD at the S155 residue were downregulated by cotreatment of deguelin and paclitaxel in SKOV3-TR cells (Figure 5C). However, RT-qPCR showed the inhibition of the expression of BCL-2 and MCL-1 was not at the transcriptional level (Figure 5D). Our results indicated that deguelin downregulated the expression of BCL-2, MCL-1, and the phosphorylation of BAD at S155 through inhibition of the EGFR signaling pathway in SKOV3-TR cells.
Deguelin Resensitized Paclitaxel in Paclitaxel-Resistant HeyA8-MDR Ovarian Cancer Cells
To investigate whether the resensitization of paclitaxel by deguelin treatment was a cell-specific phenomenon in SKOV3-TR cells, we performed a cell viability assay using HeyA8-MDR in the same experiments as above. Notably, WST-1 analysis showed that deguelin inhibited cell viability in paclitaxel-treated HeyA8-MDR cells in a dose-dependent manner (Figure 6A). The crystal violet assay also showed that deguelin inhibited cell viability in paclitaxel-treated HeyA8-MDR cells (Figure 6B). Moreover, the expression of EGFR was downregulated by cotreatment with deguelin and paclitaxel in HeyA8-MDR cells (Figure 6C). These results indicated that restoration of paclitaxel sensitivity and inhibition of EGFR by cotreatment of deguelin and paclitaxel were not cell type-specific in SKOV3-TR cells.
Discussion
As our understanding of the molecular genetics of ovarian cancer has deepened, significant progress has been made in developing effective anticancer drugs. Recently, new therapeutic approaches for ovarian cancer patients have emerged as promising options. Among these, PARP inhibitors stand as a novel class of anticancer drugs with selective lethality in BRCA1/2-mutated breast and ovarian cancers lacking homologous recombination.82 In addition to direct lethality, PARP inhibitors can activate antitumor immunity, partially through a stimulator of interferon genes (STING)-dependent mechanism.83 This effect is further enhanced by PD-1/PD-L1 blockade and occurred independently of BRCA1/2 mutation status.84 Moreover, some PARP inhibitors have demonstrated the ability to trap PARP1 and PARP2 on chromosomal DNA-damaging sites, resulting in the formation of PARP-DNA complexes.85 This phenomenon, known as “PARP trapping”, interferes with the recruitment of DNA repair enzymes,86 leading to mitotic catastrophe and subsequent cell death by aberrant DNA replication in the absence of proper DNA repair.87 This synergistic effect is observed not only with PARP inhibitors but also with alkylating agents and platinum-based agents.88 Despite numerous pharmacological studies against various cancers having been reported and clinical applications of next-generation anticancer drugs represented by targeted therapy and immunotherapy having been developed, chemical anticancer drugs, including taxane and platinum-based chemicals, are still the first-line treatment for ovarian cancer patients to date.89,90 Recently, the most important research fields against ovarian cancer have been studied on the technology of early diagnosis, the overcoming strategies of chemoresistance, and research on the ovarian cancer-specific tumor microenvironment to reveal the reason why treatment options are limited, unlike other carcinomas. Proteomics has emerged as a promising technology with the potential to address the challenges in molecular research for early diagnosis and classification of tumors for precision medicine.91 In the context of ovarian cancer, proteomic analyses offer valuable insight not only into the molecular mechanisms underlying the disease but also into the adaptive responses of tumors to therapy. Through proteomic analysis of ovarian cancer, researchers can identify potential therapeutic targets that may help mitigate chemoresistance and improve ovarian cancer patient outcomes.92 We describe here a novel paclitaxel-resensitizing capacity of deguelin against paclitaxel-resistant ovarian cancer SKOV3-TR cells.
Deguelin is a member of the rotenoid family of compounds and utilized as an insecticide.48 According to several studies, it is well known that rotenone is an inhibitor of mitochondrial NADH dehydrogenase, one of the major factors in energy metabolism, and causes cell toxicity by ATP depletion.93,94 In contrast, deguelin has been reported to selectively inhibit the growth of cancer cells by targeting oncogenic functions including cell cycle, apoptosis, and angiogenesis.48,54,95–97 For instance, it has been reported that treatment with deguelin induces apoptosis in lung cancer cells by downregulating AKT phosphorylation and stimulating ROS.98 Li et al demonstrated that deguelin inhibits cell growth in NSCLC by downregulating hexokinase 2, a key player in glycolysis metabolism.57 It has also been reported that deguelin induces cell death through the inhibition of AKT signaling and induction of the AMP-activated protein kinase (AMPK)-dependent autophagy pathway in Hep-2 head and neck cancer cells.71 Mehta et al reported that deguelin inhibits the cell growth of triple-negative breast cancer MDA-MB-231 cells by downregulating EGFR and c-Met, along with their downstream target proteins.99 Recently, Wang et al reported a study investigating the enhancement of cell cytotoxicity in triple negative-breast cancer cells by loading deguelin and paclitaxel into a nano-micelles system.100 Notably, our study is the first time that deguelin could affect paclitaxel resistance in ovarian cancer cells.
Figure 1B interestingly indicated that deguelin alone did not exert a significant effect on cell viability in SKOV3-TR cells. This raised the question of why the treatment of deguelin alone did not significantly affect the cell cytotoxicity (Figure 1B) as well as EGFR/AKT signaling (Figures 2 and 3A lane 3), and its downstream signaling (Figure 5C lane 3) in SKOV3-TR cells. Two hypotheses were considered to explain this phenomenon. Firstly, it is plausible that the sensitivity of deguelin might vary among different types of cancer cells. Second, the mechanism of action of deguelin on EGFR inhibition may be different. Gao et al indicated that deguelin directly interacts with EGFR, inhibiting its kinase activity in NSCLC cells in vitro and ex vivo.69 We compared the activation levels of EGFR in SKOV3-TR cells treated with deguelin and the EGFR-specific small molecule inhibitors gefitinib and erlotinib. As a result, gefitinib and erlotinib completely inhibited the phosphorylation of EGFR at 24 h, but deguelin did not significantly affect the phosphorylation of EGFR in SKOV3-TR cells (Supplementary Figure 1A). In addition, we demonstrated that deguelin can inhibit the activation of EGFR by EGF treatment in a dose-dependent manner in SKOV3-TR cells (Supplementary Figure 1B). This mode of action of deguelin on EGFR inhibition led us to propose that the sensitivity of EGFR, which forms various homo- or heterodimers in different cells, may inevitably vary among different cell types. Further research is underway to analyze the detailed mechanism of action of deguelin in SKOV3 cells.
We also sought to understand why the degradation of EGFR (Figure 3A lane 4) and downregulation of its downstream signaling AKT and apoptotic regulators BCL-2 and MCL-1 (Figures 2 and 5C lane 4), occurred with cotreatment with deguelin and paclitaxel rather than with deguelin alone. It has been reported that paclitaxel might affect microtubule dynamics and upregulate endocytic trafficking in cells.101 Li et al reported that paclitaxel treatment shortened the endosomal trafficking of EGFR to the perinuclear area, delivering it rapidly into the peripheral lysosomes.102 Our findings showed that the downregulation of EGFR was not significant by the treatment with deguelin alone, but the effective degradation of EGFR by the cotreatment with deguelin and paclitaxel in SKOV3-TR cells suggested that paclitaxel may enhance the lysosomal degradation of EGFR in deguelin-treated SKOV3-TR cells. Consequently, we concluded that deguelin restored paclitaxel sensitivity by promoting the lysosomal degradation of EGFR and inhibiting EGFR and its downstream signaling in SKOV3-TR cells.
Over the past decades, researchers have extensively investigated the molecular mechanisms of endocytic trafficking of EGFR and the role of the traffic in signal transduction by employing techniques such as fluorescence microscopy, receptor mutagenesis, and siRNA knockdown techniques.103–106 Accumulated evidence revealed that ligand-stimulated EGFR is recycled or degraded by receptor internalization through clathrin-mediated endocytosis (CME) with recruitment of clathrin adaptor proteins AP-2 and Grb2 or through clathrin-independent endocytosis (CIE) with the recruitment of E3 ligase c-Cbl.45,107 Under the physiological ligand environment, EGFR is rapidly recycled through the CME pathway; however, under the saturated ligand environment, EGFR is highly phosphorylated, then ubiquitinated by c-Cbl E3 ligase, and degraded through the CIE mechanism.45 The EGFR signaling pathway exerts spatial and temporal regulation to modulate various cellular processes, including proliferation, survival, differentiation, and motility, in response to environmental cues such as ligand concentration and receptor distribution. Recent studies have demonstrated that small molecules can inhibit the proliferation of cancer cells by binding to EGFR and promoting its degradation.47,108 Xiao et al reported that proguanil enhanced EGFR endocytic degradation by receptor ubiquitination in a clathrin-independent manner in bladder cancer cells.47 Yao et al reported that DPBA binds like an EGFR ligand, promotes EGFR degradation, and inhibits the proliferation of EGFR-positive NSCLC cells.46 They reported that DPBA induced EGFR degradation via the lipid raft-dependent mechanism using flotillin-1 in A549 and H1975 lung cancer cell lines.46 They also showed that DPBA does not induce degradation of EGFR via its ubiquitination. In this study, the expression level of EGFR was restored by bafilomycin A1, which inhibits lysosomal degradation, rather than the inhibitor of proteasomal degradation MG132 (Figure 3D), suggesting that deguelin can have an inhibitory mechanism of EGFR like that of DPBA, and further experiments to determine the detailed mechanism are in progress. We propose, for the first time, that deguelin promotes lysosomal degradation of EGFR, inhibits its downstream signaling, and can resensitize paclitaxel in paclitaxel-resistant ovarian cancer cells.
While many chemotherapeutic agents, including paclitaxel, are known to exert their effects through intrinsic apoptotic mechanisms, the chemoresistance mechanism of paclitaxel can vary.13,109–111 Paclitaxel binds to the N-terminal MT-loop domain of β-tubulin, affecting the dynamics of microtubule polymerization.112 It has been reported that a point mutation in the paclitaxel-binding region of β-tubulin (T26A) leads to paclitaxel resistance in cells.113 Overexpression of the ABC transporter P-gp has also been reported in various chemoresistant cancer cells.114,115 Extracellular efflux of anticancer agents, including paclitaxel and cisplatin, has been implicated in playing an important role in the chemoresistance of ovarian cancer.116 In this study, we found that deguelin did not influence the expression and function of P-gp (Figure 4). These results suggest that deguelin exerts its effects by inhibiting cell survival signaling in SKOV3-TR, leading to apoptosis induced by paclitaxel. Despite observing a significant increase in the transcriptional levels of Survivin, which functions in cancer cell survival, this upregulation did not affect the combined treatment of deguelin and paclitaxel-induced cell cytotoxicity in SKOV3-TR cells (Figures 5C and 5D). On the other hand, the effectively inhibited protein expression of BCL-2 and MCL-1 in the cotreatment implied that the expression of BCL-2 and MCL-1 was downregulated at the post-transcriptional level (Figures 5C and 5D).
AKT signaling is considered an attractive anticancer target for therapeutics. It has been known that AKT-mediated survival signals preserve various cancer cells from intrinsic and extrinsic stress.117–121 Several reports have examined the possibility that the inhibition of AKT activity can induce cell cytotoxicity by reactive oxygen species (ROS) through the suppression of the expression of ROS scavenging enzymes, including superoxide dismutase (SOD), inhibiting the FOXO signaling pathway in cancer cells.122–125 In particular, Xu et al reported that deguelin induces apoptosis through ROS by inhibiting AKT activity, and the cell cytotoxicity can be attenuated by the treatment of NAC in lung cancer cells.98 Although our results showed that the phosphorylation of AKT was inhibited by cotreatment with deguelin and paclitaxel in SKOV3-TR cells, NAC did not affect cell cytotoxicity (Figures 5A and 5B), suggesting that apoptosis induced by cotreatment with deguelin and paclitaxel in SKOV3-TR ovarian cancer cells was not caused by ROS. Since we cannot rule out the possibility of diversity in the mechanism of action of deguelin, it cannot be asserted that cotreatment with deguelin and paclitaxel does not inhibit the AKT/FOXO signaling pathway or the activation of the ROS generation mechanism in SKOV3-TR cells. The PI3K/AKT pathway is frequently altered (<70%) in ovarian cancer and contributes to aggressiveness and chemoresistance in ovarian cancer.126 It has been reported that active PI3K signaling may increase cell survival by regulating DNA damage response (DDR).127 Several studies have shown that active PI3K signaling is involved in DNA replication and cell cycle regulation, and thus, the inhibition of PI3K signaling increases replication stress and reduces the activity of the spindle assembly checkpoint protein Aurora kinase B, thereby causing mitotic catastrophe.84,127 It has also been reported that treatment with the PI3K/AKT inhibitors wortmannin and LY294002 can enhance paclitaxel-induced apoptosis in ovarian cancer cells.128 These reports showed that the function of AKT plays an important role in cell survival in response to external stress, such as anticancer drugs, and suggested that paclitaxel-induced cytotoxicity can be effectively enhanced through the regulation of the AKT downstream pathway. It has been reported that AKT can upregulate BCL-2 expression through cAMP-response element-binding protein (CREB).129 Moreover, studies have shown that the phosphorylation of BAD by AKT is involved in the paclitaxel resistance mechanism in human bladder carcinoma cells.130 Gao et al reported that deguelin downregulated MCL-1 via the AKT/GSK3β/FBW7 pathway in NSCLC cells.69 We confirmed that the expressions of BCL-2 and MCL-1 were downregulated by cotreatment with deguelin and paclitaxel in SKOV3-TR cells (Figure 5C). Although downregulations of BCL-2, MCL-1, and phosphorylation of BAD were marginally reduced by treatment with deguelin alone, it showed that cell cytotoxicity can be restored by treatment with deguelin in paclitaxel-resistant ovarian cancer cells, as the same mechanism occurs in bladder and lung cancer cells. In Figure 2, we indicated that cotreatment with deguelin and paclitaxel also inhibited the phosphorylation of STAT3, ERK, and p38 MAPK, which are common downstream signaling molecules of EGFR. Several studies have reported that STAT3,131 ERK,132 and p38 MAPK133 signaling pathways affect the proliferation of various cancer cells as well as chemoresistance, and we are analyzing the effects of these signaling pathways on proliferation and chemoresistance in ovarian cancer cells through additional studies.
Conclusion
The findings of this study underscore the potential of deguelin as an anticancer drug for the treatment of chemoresistant ovarian cancer. Our data showed that deguelin induced lysosomal degradation of EGFR, thereby inhibiting the cell survival signaling pathway. In addition, our results demonstrated that deguelin effectively resensitized the paclitaxel in SKOV3-TR cells by activating the intrinsic apoptosis pathway through downregulation of antiapoptotic factors, including BCL-2 and MCL-1. According to several studies showing the role of EGFR signaling in conferring chemoresistance to various anticancer drugs, including cisplatin in various carcinomas,26,134,135 deguelin holds promise for combating chemoresistance in EGFR-positive cancer cells.
Acknowledgments
The authors express our gratitude to the Department of Cosmetics Engineering at Konkuk University for providing support in accessing research facilities.
Author Contributions
All authors have played a crucial role in the development of this work, contributing significantly to its conception, study design, execution, data acquisition, analysis, and interpretation. Each author has actively participated in drafting, revising, and critically reviewing the article. Moreover, all authors have provided their final approval for the version to be published, unanimously selected the journal for submission, and committed to being accountable for all aspects of the work.
Funding
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) grant funded by the Korea govern (MSIT) (No. 2021R1F1A1063986).
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Lengyel E. Ovarian cancer development and metastasis. Am J Pathol. 2010;177(3):1053–1064.
2. Ghose A, Bolina A, Mahajan I, et al. Hereditary ovarian cancer: towards a cost-effective prevention strategy. Int J Environ Res Public Health. 2022;19(19). doi:10.3390/ijerph191912057
3. Schoutrop E, Moyano-Galceran L, Lheureux S, et al. Molecular, cellular and systemic aspects of epithelial ovarian cancer and its tumor microenvironment. Semi Cancer Biol. 2022;86(Pt 3):207–223. doi:10.1016/j.semcancer.2022.03.027
4. Veneziani AC, Gonzalez-Ochoa E, Alqaisi H, et al. Heterogeneity and treatment landscape of ovarian carcinoma. Nat Rev Clin Oncol. 2023;20(12):820–842. doi:10.1038/s41571-023-00819-1
5. Handley KF, Sims TT, Bateman NW, et al. Classification of high-grade serous ovarian cancer using tumor morphologic characteristics. JAMA network open. 2022;5(10):e2236626. doi:10.1001/jamanetworkopen.2022.36626
6. Pavlik EJ, Smith C, Dennis TS, et al. Disease-specific survival of type I and type II epithelial ovarian cancers-stage challenges categorical assignments of indolence & aggressiveness. Diagnostics. 2020;10(2). doi:10.3390/diagnostics10020056
7. Barnes BM, Nelson L, Tighe A, et al. Distinct transcriptional programs stratify ovarian cancer cell lines into the five major histological subtypes. Genome Med. 2021;13(1):140. doi:10.1186/s13073-021-00952-5
8. Shu C, Zheng X, Wuhafu A, et al. Acquisition of taxane resistance by p53 inactivation in ovarian cancer cells. Acta Pharmacol. Sin. 2022;43(9):2419–2428. doi:10.1038/s41401-021-00847-6
9. Tendulkar S, Dodamani S. Chemoresistance in ovarian cancer: prospects for new drugs. Anti Cancer Agent Med Chem. 2021;21(6):668–678. doi:10.2174/1871520620666200908104835
10. Ghose A, McCann L, Makker S, et al. Diagnostic biomarkers in ovarian cancer: advances beyond CA125 and HE4. Therapeut Adv Med Oncol. 2024;16:17588359241233225. doi:10.1177/17588359241233225
11. Ahmed Khalil A, Rauf A, Alhumaydhi FA, et al. Recent developments and anticancer therapeutics of paclitaxel: an update. Curr. Pharm. Des. 2022;28(41):3363–3373. doi:10.2174/1381612829666221102155212
12. Mosca L, Ilari A, Fazi F, Assaraf YG, Colotti G. Taxanes in cancer treatment: activity, chemoresistance and its overcoming. Drug Resist Updat. 2021;54:100742. doi:10.1016/j.drup.2020.100742
13. Zhao S, Tang Y, Wang R, Najafi M. Mechanisms of cancer cell death induction by paclitaxel: an updated review. Apoptosis. 2022;27(9–10):647–667. doi:10.1007/s10495-022-01750-z
14. Faria RS, De lima LI, Bonadio RS, et al. Liposomal paclitaxel induces apoptosis, cell death, inhibition of migration capacity and antitumoral activity in ovarian cancer. Biomed Pharmacothe. 2021;142:112000. doi:10.1016/j.biopha.2021.112000
15. Jaunky DB, Larocque K, Husser MC, Liu JT, Forgione P, Piekny A. Characterization of a recently synthesized microtubule-targeting compound that disrupts mitotic spindle Poles in human cells. Sci Rep. 2021;11(1):23665. doi:10.1038/s41598-021-03076-3
16. Sazonova EV, Petrichuk SV, Kopeina GS, Zhivotovsky B. A link between mitotic defects and mitotic catastrophe: detection and cell fate. Biology Direct. 2021;16(1):25. doi:10.1186/s13062-021-00313-7
17. Drosos Y, Konstantakou EG, Bassogianni AS, et al. Microtubule dynamics deregulation induces apoptosis in human urothelial bladder cancer cells via a p53-independent pathway. Cancers. 2023;15(14):2.
18. Ren X, Zhao B, Chang H, Xiao M, Wu Y, Liu Y. Paclitaxel suppresses proliferation and induces apoptosis through regulation of ROS and the AKT/MAPK signaling pathway in canine mammary gland tumor cells. Mol Med Rep. 2018;17(6):8289–8299. doi:10.3892/mmr.2018.8868
19. Pan Z, Avila A, Gollahon L. Paclitaxel induces apoptosis in breast cancer cells through different calcium--regulating mechanisms depending on external calcium conditions. Int J Mol Sci. 2014;15(2):2672–2694. doi:10.3390/ijms15022672
20. Miller AV, Hicks MA, Nakajima W, Richardson AC, Windle JJ, Harada H. Paclitaxel-induced apoptosis is BAK-dependent, but BAX and BIM-independent in breast tumor. PLoS One. 2013;8(4):e60685. doi:10.1371/journal.pone.0060685
21. Zhang X, Wu X, Zhang F, et al. Paclitaxel induces apoptosis of esophageal squamous cell carcinoma cells by downregulating STAT3 phosphorylation at Ser727. Oncol Rep. 2017;37(4):2237–2244. doi:10.3892/or.2017.5503
22. He Y, Sun MM, Zhang GG, et al. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct Target Ther. 2021;6(1):425. doi:10.1038/s41392-021-00828-5
23. Levantini E, Maroni G, Del Re M, Tenen DG. EGFR signaling pathway as therapeutic target in human cancers. Semi Cancer Biol. 2022;85:253–275. doi:10.1016/j.semcancer.2022.04.002
24. Rosenkranz AA, Slastnikova TA. Epidermal growth factor receptor: key to selective intracellular delivery. Biochem Biokhimiia. 2020;85(9):967–1092. doi:10.1134/s0006297920090011
25. Bai X, Sun P, Wang X, et al. Structure and dynamics of the EGFR/HER2 heterodimer. Cell Discovery. 2023;9(1):18. doi:10.1038/s41421-023-00523-5
26. Uribe ML, Marrocco I, Yarden Y. EGFR in cancer: signaling mechanisms, drugs, and acquired resistance. Cancers. 2021;13(11):3.
27. Lipsick J. A history of cancer research: tyrosine kinases. Cold Spring Harbor Perspect Biol. 2019;11(2):3.
28. Xu H, Zong H, Ma C, et al. Epidermal growth factor receptor in glioblastoma. Oncol Lett. 2017;14(1):512–516. doi:10.3892/ol.2017.6221
29. Bethune G, Bethune D, Ridgway N, Xu Z. Epidermal growth factor receptor (EGFR) in lung cancer: an overview and update. J Thorac Dis. 2010;2(1):48–51.
30. Spano JP, Lagorce C, Atlan D, et al. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann Oncol. 2005;16(1):102–108. doi:10.1093/annonc/mdi006
31. Rehmani HS, Issaeva N. EGFR in head and neck squamous cell carcinoma: exploring possibilities of novel drug combinations. Ann Transl Med. 2020;8(13):813. doi:10.21037/atm.2020.04.07
32. Oliveira-Cunha M, Newman WG, Siriwardena AK. Epidermal growth factor receptor in pancreatic cancer. Cancers. 2011;3(2):1513–1526. doi:10.3390/cancers3021513
33. Masuda H, Zhang D, Bartholomeusz C, Doihara H, Hortobagyi GN, Ueno NT. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat. 2012;136(2):331–345. doi:10.1007/s10549-012-2289-9
34. Thakur A, Faujdar C, Sharma R, et al. Glioblastoma: current status, emerging targets, and recent advances. J Med Chem. 2022;65(13):8596–8685. doi:10.1021/acs.jmedchem.1c01946
35. Smolle E, Leithner K, Olschewski H. Oncogene addiction and tumor mutational burden in non-small-cell lung cancer: clinical significance and limitations. Thorac Cancer. 2020;11(2):205–215. doi:10.1111/1759-7714.13246
36. Amisha F, Malik P, Saluja P, et al. A comprehensive review on the role of human epidermal growth factor receptor 2 (HER2) as a biomarker in extra-mammary and extra-gastric cancers. Onco. 2023;3(2):96–124.
37. Sheng Q, Liu J. The therapeutic potential of targeting the EGFR family in epithelial ovarian cancer. Br. J. Cancer. 2011;104(8):1241–1245.
38. Mehner C, Oberg AL, Goergen KM, et al. EGFR as a prognostic biomarker and therapeutic target in ovarian cancer: evaluation of patient cohort and literature review. Genes Cancer. 2017;8(5–6):589–599. doi:10.18632/genesandcancer.142
39. Forlani L, De Cecco L, Simeon V, et al. Biological and clinical impact of membrane EGFR expression in a subgroup of OC patients from the Phase IV ovarian cancer MITO-16A/Mango-OV2A trial. J Experiment Clin Cancer Res. 2023;42(1):83. doi:10.1186/s13046-023-02651-y
40. Schultz DF, Billadeau DD, Jois SD. EGFR trafficking: effect of dimerization, dynamics, and mutation. Front Oncol. 2023;13:1258371. doi:10.3389/fonc.2023.1258371
41. Bakker J, Spits M, Neefjes J, Berlin I. The EGFR odyssey - from activation to destruction in space and time. J Cell Sci. 2017;130(24):4087–4096. doi:10.1242/jcs.209197
42. Melikova MS, Kondratov KA, Kornilova ES. Two different stages of epidermal growth factor (EGF) receptor endocytosis are sensitive to free ubiquitin depletion produced by proteasome inhibitor MG132. Cell Biol Int. 2006;30(1):31–43. doi:10.1016/j.cellbi.2005.09.003
43. Longva KE, Blystad FD, Stang E, Larsen AM, Johannessen LE, Madshus IH. Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J Cell Biol. 2002;156(5):843–854. doi:10.1083/jcb.200106056
44. Oksvold MP, Skarpen E, Wierod L, Paulsen RE, Huitfeldt HS. Re-localization of activated EGF receptor and its signal transducers to multivesicular compartments downstream of early endosomes in response to EGF. Eur J Cell Biol. 2001;80(4):285–294. doi:10.1078/0171-9335-00160
45. Zhou Y, Sakurai H. New trend in ligand-induced EGFR trafficking: a dual-mode clathrin-mediated endocytosis model. J Proteom. 2022;255:104503. doi:10.1016/j.jprot.2022.104503
46. Yao N, Wang C-R, Liu M-Q, et al. Discovery of a novel EGFR ligand DPBA that degrades EGFR and suppresses EGFR-positive NSCLC growth. Sig Transd Targ Ther. 2020;5(1):1–13.
47. Xiao D, Hu X, Peng M, et al. Inhibitory role of proguanil on the growth of bladder cancer via enhancing EGFR degradation and inhibiting its downstream signaling pathway to induce autophagy. Cell Death Dis. 2022;13(5):499. doi:10.1038/s41419-022-04937-z
48. Lin ZY, Yun QZ, Wu L, Zhang TW, Yao TZ. Pharmacological basis and new insights of deguelin concerning its anticancer effects. Pharmacol Res. 2021;174:105935. doi:10.1016/j.phrs.2021.105935
49. Zhang P, Zhang M, Mellich TA, Pearson BJ, Chen J, Zhang Z. Variation in rotenone and deguelin contents among strains across four tephrosia species and their activities against aphids and whiteflies. Toxins. 2022;14(5):3.
50. Ibarra-Gutiérrez MT, Serrano-García N, Orozco-Ibarra M. Rotenone-induced model of parkinson’s disease: beyond mitochondrial complex I inhibition. Molec Neurobiol. 2023;60(4):1929–1948. doi:10.1007/s12035-022-03193-8
51. Thirugnanam T, Santhakumar K. Chemically induced models of Parkinson’s disease. Comp Biochem Physiol Toxicol Pharmacol. 2022;252:109213. doi:10.1016/j.cbpc.2021.109213
52. Boyd J, Han A. Deguelin and its role in chronic diseases. Adv Exp Med Biol. 2016;929:363–375. doi:10.1007/978-3-319-41342-6_16
53. Baba Y, Kato Y. Deguelin, a novel anti-tumorigenic agent in human esophageal squamous cell carcinoma. EBioMedicine. 2017;26:10. doi:10.1016/j.ebiom.2017.11.010
54. Wang Y, Ma W, Zheng W. Deguelin, a novel anti-tumorigenic agent targeting apoptosis, cell cycle arrest and anti-angiogenesis for cancer chemoprevention. Mol Clin Oncol. 2013;1(2):215–219. doi:10.3892/mco.2012.36
55. Hu J, Ye H, Fu A, et al. Deguelin--an inhibitor to tumor lymphangiogenesis and lymphatic metastasis by downregulation of vascular endothelial cell growth factor-D in lung tumor model. Int, J, Cancer. 2010;127(10):2455–2466. doi:10.1002/ijc.25253
56. Henrich CJ, Cartner LK, Wilson JA, et al. Deguelins, natural product modulators of NF1-defective astrocytoma cell growth identified by high-throughput screening of partially purified natural product extracts. J Nat Prod. 2015;78(11):2776–2781. doi:10.1021/acs.jnatprod.5b00753
57. Li W, Gao F, Ma X, Wang R, Dong X, Wang W. Deguelin inhibits non-small cell lung cancer via down-regulating Hexokinases II-mediated glycolysis. Oncotarget. 2017;8(20):32586.
58. Li M, Yu X, Li W, et al. Deguelin suppresses angiogenesis in human hepatocellular carcinoma by targeting HGF-c-Met pathway. Oncotarget. 2018;9(1):152.
59. Chen L, Jiang K, Chen H, et al. Deguelin induces apoptosis in colorectal cancer cells by activating the p38 MAPK pathway. Cancer Manage Res. 2019;11:95.
60. Yu X, Liang Q, Liu W, Zhou L, Li W, Liu H. Deguelin, an Aurora B kinase inhibitor, exhibits potent anti-tumor effect in human esophageal squamous cell carcinoma. EBioMedicine. 2017;26:100–111.
61. Carpenter EL, Chagani S, Nelson D, et al. Mitochondrial complex I inhibitor deguelin induces metabolic reprogramming and sensitizes vemurafenib‐resistant BRAFV600E mutation bearing metastatic melanoma cells. Molec Carcinogene. 2019;58(9):1680–1690.
62. Kim HS, Hoang V-H, Hong M, et al. Investigation of B, C-ring truncated deguelin derivatives as heat shock protein 90 (HSP90) inhibitors for use as anti-breast cancer agents. Bioorg. Med. Chem. 2019;27(7):1370–1381.
63. Varughese RS, Lam WST, Marican AA, et al. Biopharmacological considerations for accelerating drug development of deguelin, a rotenoid with potent chemotherapeutic and chemopreventive potential. Cancer. 2019;125(11):1789–1798.
64. Li W, Yu X, Xia Z, et al. Repression of Noxa by Bmi1 contributes to deguelin‐induced apoptosis in non‐small cell lung cancer cells. J Cell & Mol Med. 2018;22(12):6213–6227.
65. Li W, Yu X, Ma X, et al. Deguelin attenuates non-small cell lung cancer cell metastasis through inhibiting the CtsZ/FAK signaling pathway. Cell. Signalling. 2018;50:131–141.
66. Martincuks A, Li PC, Zhao Q, et al. CD44 in ovarian cancer progression and therapy resistance-A critical role for STAT3. Front Oncol. 2020;10:589601. doi:10.3389/fonc.2020.589601
67. Salaroglio IC, Mungo E, Gazzano E, Kopecka J, Riganti C. ERK is a Pivotal Player of Chemo-Immune-Resistance in Cancer. Int J Mol Sci. 2019;20:2.
68. Liu YP, Zheng CC, Huang YN, He ML, Xu WW, Li B. Molecular mechanisms of chemo- and radiotherapy resistance and the potential implications for cancer treatment. MedComm. 2021;2(3):315–340. doi:10.1002/mco2.55
69. Gao F, Yu X, Li M, et al. Deguelin suppresses non-small cell lung cancer by inhibiting EGFR signaling and promoting GSK3beta/FBW7-mediated Mcl-1 destabilization. Cell Death Dis. 2020;11(2):143. doi:10.1038/s41419-020-2344-0
70. Murillo G, Peng X, Torres KE, Mehta RG. Deguelin inhibits growth of breast cancer cells by modulating the expression of key members of the Wnt signaling pathway. Cancer Prev Res. 2009;2(11):942–950. doi:10.1158/1940-6207.CAPR-08-0232
71. Yang YL, Ji C, Bi ZG, et al. Deguelin induces both apoptosis and autophagy in cultured head and neck squamous cell carcinoma cells. PLoS One. 2013;8(1):e54736. doi:10.1371/journal.pone.0054736
72. Pote MS, Gacche RN. ATP-binding cassette efflux transporters and MDR in cancer. Drug Discovery Today. 2023;28(5):103537. doi:10.1016/j.drudis.2023.103537
73. Pilotto Heming C, Muriithi W, Wanjiku Macharia L, Niemeyer Filho P, Moura-Neto V, Aran V. P-glycoprotein and cancer: what do we currently know? Heliyon. 2022;8(10):e11171. doi:10.1016/j.heliyon.2022.e11171
74. Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene. 2003;22(47):7468–7485. doi:10.1038/sj.onc.1206948
75. Mirzaei S, Gholami MH, Hashemi F, et al. Advances in understanding the role of P-gp in doxorubicin resistance: molecular pathways, therapeutic strategies, and prospects. Drug Discovery Today. 2022;27(2):436–455. doi:10.1016/j.drudis.2021.09.020
76. Orlicky J, Sulova Z, Dovinova I, Fiala R, Zahradnikova A, Breier A. Functional fluo-3/AM assay on P-glycoprotein transport activity in L1210/VCR cells by confocal microscopy. Gen Physiol Biophys. 2004;23(3):357–366.
77. Liang X, Huang Y. Intracellular free calcium concentration and cisplatin resistance in human lung adenocarcinoma A549 cells. Biosci Rep. 2000;20(3):129–138. doi:10.1023/a:1005530501137
78. Zhang Y, Tang Y, Tang X, Wang Y, Zhang Z, Yang H. Paclitaxel induces the apoptosis of prostate cancer cells via ROS-mediated HIF-1alpha expression. Molecules. 2022;27(21):3.
79. Glaviano A, Foo ASC, Lam HY, et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer. 2023;22(1):138. doi:10.1186/s12943-023-01827-6
80. Koundouros N, Poulogiannis G. Phosphoinositide 3-Kinase/Akt Signaling and redox metabolism in cancer. Front Oncol. 2018;8:160. doi:10.3389/fonc.2018.00160
81. Liu R, Chen Y, Liu G, et al. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020;11(9):797. doi:10.1038/s41419-020-02998-6
82. Luo L, Keyomarsi K. PARP inhibitors as single agents and in combination therapy: the most promising treatment strategies in clinical trials for BRCA-mutant ovarian and triple-negative breast cancers. Expert Opin Invest Drugs. 2022;31(6):607–631. doi:10.1080/13543784.2022.2067527
83. Ding L, Kim HJ, Wang Q, et al. PARP inhibition elicits STING-dependent antitumor immunity in brca1-deficient ovarian cancer. Cell Rep. 2018;25(11):2972–2980.e5. doi:10.1016/j.celrep.2018.11.054
84. Aliyuda F, Moschetta M, Ghose A, et al. Advances in ovarian cancer treatment beyond PARP inhibitors. Curr Cancer Drug Targets. 2023;23(6):433–446. doi:10.2174/1568009623666230209121732
85. Mahadevan J, Jha A, Rudolph J, et al. Dynamics of endogenous PARP1 and PARP2 during DNA damage revealed by live-cell single-molecule imaging. iScience. 2023;26(1):105779. doi:10.1016/j.isci.2022.105779
86. Zheng F, Zhang Y, Chen S, Weng X, Rao Y, Fang H. Mechanism and current progress of Poly ADP-ribose polymerase (PARP) inhibitors in the treatment of ovarian cancer. Biomed Pharmacother. 2020;123:109661. doi:10.1016/j.biopha.2019.109661
87. Li Q, Qian W, Zhang Y, Hu L, Chen S, Xia Y. A new wave of innovations within the DNA damage response. Signal Transduct Target Ther. 2023;8(1):338. doi:10.1038/s41392-023-01548-8
88. Wang M, Chen S, Ao D. Targeting DNA repair pathway in cancer: mechanisms and clinical application. MedComm. 2021;2(4):654–691. doi:10.1002/mco2.103
89. Patel A, Kalachand R, Busschots S, et al. Taxane monotherapy regimens for the treatment of recurrent epithelial ovarian cancer. Cochrane Database Syst Rev. 2022;7(7):Cd008766. doi:10.1002/14651858.CD008766.pub3
90. Garrido MP, Fredes AN, Lobos-González L, Valenzuela-Valderrama M, Vera DB, Romero C. Current treatments and new possible complementary therapies for epithelial ovarian cancer. Biomedicines. 2021;10:1.
91. Su M, Zhang Z, Zhou L, Han C, Huang C, Nice EC. Proteomics, personalized medicine and cancer. Cancers. 2021;13(11). doi:10.3390/cancers13112512
92. Ghose A, Gullapalli SVN, Chohan N, et al. Applications of proteomics in ovarian cancer: dawn of a new era. Proteomes. 2022;10(2):2.
93. Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J Biol Chem. 1992;267(8):5317–5323.
94. Singer TP, Ramsay RR. The reaction sites of rotenone and ubiquinone with mitochondrial NADH dehydrogenase. Biochim Biophys Acta. 1994;1187(2):198–202. doi:10.1016/0005-2728(94)90110-4
95. Kang W, Zheng X, Wang P, Guo S. Deguelin exerts anticancer activity of human gastric cancer MGC-803 and MKN-45 cells in vitro. IntJ Mol Med. 2018;41(6):3157–3166. doi:10.3892/ijmm.2018.3532
96. Tuli HS, Mittal S, Loka M, et al. Deguelin targets multiple oncogenic signaling pathways to combat human malignancies. Pharmacol Res. 2021;166:105487. doi:10.1016/j.phrs.2021.105487
97. Lokhande KB, Nagar S, Swamy KV. Molecular interaction studies of Deguelin and its derivatives with Cyclin D1 and Cyclin E in cancer cell signaling pathway: the computational approach. Sci Rep. 2019;9(1):1778. doi:10.1038/s41598-018-38332-6
98. Xu H, Li X, Ding W, et al. Deguelin induces the apoptosis of lung cancer cells through regulating a ROS driven Akt pathway. Cancer Cell Int. 2015;15:25. doi:10.1186/s12935-015-0166-4
99. Mehta R, Katta H, Alimirah F, et al. Deguelin action involves c-Met and EGFR signaling pathways in triple negative breast cancer cells. PLoS One. 2013;8(6):e65113. doi:10.1371/journal.pone.0065113
100. Wang Y, Lan Y, Wu L, Zhang S, Su Q, Yang Q. Deguelin and paclitaxel loaded PEG-PCL nano-micelles for suppressing the proliferation and inducing apoptosis of breast cancer cells. Front Biosci. 2024;29(2):90. doi:10.31083/j.fbl2902090
101. Wordeman L, Vicente JJ. Microtubule targeting agents in disease: classic drugs, novel roles. Cancers. 2021;13(22):2.
102. Li H, Duan ZW, Xie P, et al. Effects of paclitaxel on EGFR endocytic trafficking revealed using quantum dot tracking in single cells. PLoS One. 2012;7(9):e45465. doi:10.1371/journal.pone.0045465
103. Martin-Fernandez ML, Clarke DT, Tobin MJ, Jones GR. Real-time studies of the interactions between epidermal growth factor and its receptor during endocytic trafficking. Cell Mol Biol. 2000;46(6):1103–1112.
104. Hampton KK, Craven RJ. Pathways driving the endocytosis of mutant and wild-type EGFR in cancer. Oncoscience. 2014;1(8):504–512. doi:10.18632/oncoscience.67
105. Tanaka T, Zhou Y, Ozawa T, et al. Ligand-activated epidermal growth factor receptor (EGFR) signaling governs endocytic trafficking of unliganded receptor monomers by non-canonical phosphorylation. J Biol Chem. 2018;293(7):2288–2301. doi:10.1074/jbc.M117.811299
106. Rush JS, Quinalty LM, Engelman L, Sherry DM, Ceresa BP. Endosomal accumulation of the activated epidermal growth factor receptor (EGFR) induces apoptosis. J Biol Chem. 2012;287(1):712–722. doi:10.1074/jbc.M111.294470
107. Papagiannouli F. Endocytosis at the crossroad of polarity and signaling regulation: learning from drosophila melanogaster and beyond. Int J Mole Sci. 2022;23(9):4.
108. Yao N, Wang CR, Liu MQ, et al. Discovery of a novel EGFR ligand DPBA that degrades EGFR and suppresses EGFR-positive NSCLC growth. Sig Transd Targ Ther. 2020;5(1):214. doi:10.1038/s41392-020-00251-2
109. Cui H, Arnst K, Miller DD, Li W. Recent advances in elucidating paclitaxel resistance mechanisms in non-small cell lung cancer and strategies to overcome drug resistance. Curr Med Chem. 2020;27(39):6573–6595. doi:10.2174/0929867326666191016113631
110. Das T, Anand U, Pandey SK, et al. Therapeutic strategies to overcome taxane resistance in cancer. Drug Resist Updates. 2021;55:100754. doi:10.1016/j.drup.2021.100754
111. Nunes M, Silva PMA, Coelho R, et al. Generation of two paclitaxel-resistant high-grade serous carcinoma cell lines with increased expression of p-glycoprotein. Front Oncol. 2021;11:752127. doi:10.3389/fonc.2021.752127
112. Prota AE, Lucena-Agell D, Ma Y, et al. Structural insight into the stabilization of microtubules by taxanes. eLife. 2023:12. doi:10.7554/eLife.84791
113. Hari M, Loganzo F, Annable T, et al. Paclitaxel-resistant cells have a mutation in the paclitaxel-binding region of beta-tubulin (Asp26Glu) and less stable microtubules. Mol Cancer Ther. 2006;5(2):270–278. doi:10.1158/1535-7163.MCT-05-0190
114. Xiao H, Zheng Y, Ma L, Tian L, Sun Q. Clinically-relevant ABC transporter for anti-cancer drug resistance. Front Pharmacol. 2021;12:648407. doi:10.3389/fphar.2021.648407
115. Tian Y, Lei Y, Wang Y, Lai J, Wang J, Xia F. Mechanism of multidrug resistance to chemotherapy mediated by P‑glycoprotein (Review). Int j Oncol. 2023;63:5.
116. Alatise KL, Gardner S, Alexander-Bryant A. Mechanisms of drug resistance in ovarian cancer and associated gene targets. Cancers. 2022;14(24). doi:10.3390/cancers14246246
117. Los M, Maddika S, Erb B, Schulze-Osthoff K. Switching Akt: from survival signaling to deadly response. Bioessays. 2009;31(5):492–495. doi:10.1002/bies.200900005
118. Kennedy SG, Wagner AJ, Conzen SD, et al. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997;11(6):701–713. doi:10.1101/gad.11.6.701
119. Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96(6):857–868. doi:10.1016/s0092-8674(00)80595-4
120. Adil MS, Khulood D, Somanath PR. Targeting Akt-associated microRNAs for cancer therapeutics. Biochem. Pharmacol. 2021;189:114384. doi:10.1016/j.bcp.2020.114384
121. Mundi PS, Sachdev J, McCourt C, Kalinsky K. AKT in cancer: new molecular insights and advances in drug development. Br J Clin Pharmacol. 2016;82(4):943–956. doi:10.1111/bcp.13021
122. Nasimian A, Farzaneh P, Tamanoi F, Bathaie SZ. Cytosolic and mitochondrial ROS production resulted in apoptosis induction in breast cancer cells treated with Crocin: the role of FOXO3a, PTEN and AKT signaling. Biochem Pharmacol. 2020;177:113999. doi:10.1016/j.bcp.2020.113999
123. Hayes JD, Dinkova-Kostova AT, Tew KD. Oxidative Stress in Cancer. Cancer Cell. 2020;38(2):167–197. doi:10.1016/j.ccell.2020.06.001
124. Perillo B, Di Donato M, Pezone A, et al. ROS in cancer therapy: the bright side of the moon. Experiment Mole Med. 2020;52(2):192–203. doi:10.1038/s12276-020-0384-2
125. Nakamura H, Takada K. Reactive oxygen species in cancer: current findings and future directions. Cancer Sci. 2021;112(10):3945–3952. doi:10.1111/cas.15068
126. Ghoneum A, Said N. PI3K-AKT-mTOR and NFκB pathways in ovarian cancer: implications for targeted therapeutics. Cancers. 2019;11:7.
127. Huang TT, Lampert EJ, Coots C, Lee JM. Targeting the PI3K pathway and DNA damage response as a therapeutic strategy in ovarian cancer. Cancer Treat Rev. 2020;86:102021. doi:10.1016/j.ctrv.2020.102021
128. Rinne N, Christie EL, Ardasheva A, et al. Targeting the PI3K/AKT/mTOR pathway in epithelial ovarian cancer, therapeutic treatment options for platinum-resistant ovarian cancer. Cancer Drug Resist. 2021;4(3):573–595. doi:10.20517/cdr.2021.05
129. Steven A, Friedrich M, Jank P, et al. What turns CREB on? And off? And why does it matter? Cell Mol Life Sci. 2020;77(20):4049–4067. doi:10.1007/s00018-020-03525-8
130. Szanto A, Bognar Z, Szigeti A, Szabo A, Farkas L, Gallyas F. Critical role of bad phosphorylation by Akt in cytostatic resistance of human bladder cancer cells. Anticancer Res. 2009;29(1):159–164.
131. Dong J, Cheng XD, Zhang WD, Qin JJ. Recent update on development of small-molecule STAT3 inhibitors for cancer therapy: from phosphorylation inhibition to protein degradation. J Med Chem. 2021;64(13):8884–8915. doi:10.1021/acs.jmedchem.1c00629
132. Chesnokov MS, Khan I, Park Y, et al. The MEK1/2 pathway as a therapeutic target in high-grade serous ovarian carcinoma. Cancers. 2021;13(6):2.
133. Kudaravalli S, den Hollander P, Mani SA. Role of p38 MAP kinase in cancer stem cells and metastasis. Oncogene. 2022;41(23):3177–3185. doi:10.1038/s41388-022-02329-3
134. Li A, Cao W, Liu X, et al. Gefitinib sensitization of cisplatin-resistant wild-type EGFR non-small cell lung cancer cells. J Cancer Res Clin Oncol. 2020;146(7):1737–1749. doi:10.1007/s00432-020-03228-4
135. Lei ZN, Tian Q, Teng QX, et al. Understanding and targeting resistance mechanisms in cancer. MedComm. 2023;4(3):e265. doi:10.1002/mco2.265
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.