")
Back to Journals » International Journal of Nanomedicine » Volume 19
Multimodal, PSMA-Targeted, PAMAM Dendrimer-Drug Conjugates for Treatment of Prostate Cancer: Preclinical Evaluation
Authors Lesniak WG , Boinapally S, Lofland G, Jiang Z, Foss CA, Behman Azad B, Jablonska A, Garcia MA, Brzezinski M, Pomper MG
Received 9 December 2023
Accepted for publication 28 March 2024
Published 30 May 2024 Volume 2024:19 Pages 4995—5010
DOI https://doi.org/10.2147/IJN.S454128
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Kamakhya Misra
Wojciech G Lesniak,1 Srikanth Boinapally,1 Gabriela Lofland,1 Zirui Jiang,1 Catherine A Foss,1 Babak Behman Azad,1 Anna Jablonska,1,2 Mauro A Garcia,1 Maria Brzezinski,1 Martin G Pomper1,3
1Russell H. Morgan Department of Radiology and Radiological Science, Johns Hopkins University, Baltimore, MD, 21287, USA; 2Institute for Cell Engineering, Johns Hopkins University, Baltimore, MD, 21218, USA; 3Department of Radiology, University of Texas Southwestern Medical Center, Dallas, TX, 75390, USA
Correspondence: Wojciech G Lesniak, Russell H. Morgan Department of Radiology and Radiological Science, Johns Hopkins University, 600 N. Wolfe Street, Baltimore, MD, 21287, USA, Email [email protected]
Introduction: Prostate cancer (PC) is the second most common cancer and the fifth most frequent cause of cancer death among men. Prostate-specific membrane antigen (PSMA) expression is associated with aggressive PC, with expression in over 90% of patients with metastatic disease. Those characteristics have led to its use for PC diagnosis and therapies with radiopharmaceuticals, antibody-drug conjugates, and nanoparticles. Despite these advancements, none of the current therapeutics are curative and show some degree of toxicity. Here we present the synthesis and preclinical evaluation of a multimodal, PSMA-targeted dendrimer-drug conjugate (PT-DDC), synthesized using poly(amidoamine) (PAMAM) dendrimers. PT-DDC was designed to enable imaging of drug delivery, providing valuable insights to understand and enhance therapeutic response.
Methods: The PT-DDC was synthesized through consecutive conjugation of generation-4 PAMAM dendrimers with maytansinoid-1 (DM1) a highly potent antimitotic agent, Cy5 infrared dye for optical imaging, 2,2′,2”-(1,4,7-triazacyclononane-1,4,7-triyl)triacetic acid (NOTA) chelator for radiolabeling with copper-64 and positron emission tomography tomography/computed tomography (PET/CT), lysine-urea-glutamate (KEU) PSMA-targeting moiety and the remaining terminal primary amines were capped with butane-1,2-diol. Non-targeted control dendrimer-drug conjugate (Ctrl-DDC) was formulated without conjugation of KEU. PT-DDC and Ctrl-DDC were characterized using high-performance liquid chromatography, matrix assisted laser desorption ionization mass spectrometry and dynamic light scattering. In vitro and in vivo evaluation of PT-DDC and Ctrl-DDC were carried out in isogenic human prostate cancer PSMA+ PC3 PIP and PSMA− PC3 flu cell lines, and in mice bearing the corresponding xenografts.
Results: PT-DDC was stable in 1×PBS and human blood plasma and required glutathione for DM1 release. Optical, PET/CT and biodistribution studies confirmed the in vivo PSMA-specificity of PT-DDC. PT-DDC demonstrated dose-dependent accumulation and cytotoxicity in PSMA+ PC3 PIP cells, and also showed growth inhibition of the corresponding tumors. PT-DDC did not accumulate in PSMA− PC3 flu tumors and did not inhibit their growth. Ctrl-DDC did not show PSMA specificity.
Conclusion: In this study, we synthesized a multimodal theranostic agent capable of delivering DM1 and a radionuclide to PSMA+ tumors. This approach holds promise for enhancing image-guided treatment of aggressive, metastatic subtypes of prostate cancer.
Keywords: prostate cancer, PSMA, PAMAM dendrimer, targeted cancer treatment, theranostics
Graphical Abstract:
Introduction
Prostate cancer (PC) is the most frequently diagnosed non-cutaneous cancer and the second most common cause of cancer-related death in men in the United States.1 Patients with metastatic, castration-resistant PC (mCRPC) experience significant debilitation and often require aggressive treatments, negatively affecting their quality of life. Early detection of aggressive PC and the attendant earlier selection of appropriate treatment increases the survival rate.2–4 Non-invasive molecular imaging and image-guided therapies are being increasingly employed in cancer management. Imaging of prostate-specific membrane antigen (PSMA) expression is being evaluated extensively, as it is overexpressed in most PCs, enabling their detection and treatment.5–7 PSMA has been leveraged for diagnosis, radiopharmaceutical therapy and targeted drug delivery by low-molecular-weight agents, antibody-drug conjugates (ADCs) and polylactic acid-polyethylene glycol (PLA-PEG)-based BIND-014 nanoparticles.5,6,8–12 Positron emission tomography/computed tomography (PET/CT) imaging with PSMA-specific radiotracers, such as recently approved [18F]DCFPyL (PYLARIFY®), is the preferred imaging modality for staging and evaluating patients with biochemical recurrence.6 Successful application of low-molecular-weight PSMA-specific PET/CT radiotracers in PC diagnosis has triggered significant effort aimed at developing lysine-urea-glutamate (KEU)-based radiotherapeutics equipped with β- or α-particle-emitting radionuclides.11 Recently, PSMA-targeted, lutetium-177 vipivotide tetraxetan (PLUVICTO®) has been approved for mCRPC. Although PLUVICTO® has demonstrated a marked increase in quality of life, it conveys only a modest survival benefit.13 Clinical evaluation of MLN2704 and PSMA-ADC, ADCs conjugated with potent antimitotic agents DM1 and monomethyl auristatin E (MMAE), respectively, proved the feasibility of PC treatment with ADCs. However, MLN2704 and PSMA-ADC caused a peripheral neuropathy at therapeutic doses.14,15 PSMA-targeted BIND-014 nanoparticles loaded with docetaxel displayed moderate efficacy in patients with PSMA-positive as well as negative cancers.16,17 That lack of PSMA specificity and modest efficacy may be related to the relatively large size of BIND-014 nanoparticles (~50 nm), leading mainly to passive cancer accumulation due to the enhanced permeability and retention (EPR) effect.18 Inspired by results obtained for PSMA-targeted radiopharmaceuticals, ADCs and BIND-014, we have developed PSMA-targeted poly(amidoamine) (PAMAM) dendrimers for active PC targeting with a potential for highly specific delivery of imaging and therapeutic agents.19,20 PAMAM dendrimers are significantly smaller than ADCs and BIND-014, predisposing them to improved pharmacokinetics, permeability through biological barriers and active cancer targeting. PAMAM dendrimers are chemically and biologically stable, have many reactive terminal groups and bulky internal void volume, and their size and composition can easily be tuned depending on the application.19–22 PAMAM dendrimer-drug conjugates recently featured in several clinical trials for treatment of neovascular age-related macular degeneration, diabetic macular edema, COVID-19, and liver cancer. [https://clinicaltrials.gov] We have shown in previous work that generation-4 PAMAM dendrimers conjugated with KEU and several other functionalities including chelating agents and fluorescent dyes showed significantly higher uptake in PSMA+ PC3 PIP tumors vs PSMA− PC3 flu tumors with low uptake in off-target tissues.19 In the following studies, we demonstrated that our PSMA-targeted PAMAM dendrimers conjugated with multiple Cy7.5 near infrared (NIR) dyes enabled imaging of PSMA expression with NIR optical and photoacoustic (PA) imaging with superior distribution and cellular internalization in PSMA+ PC3 PIP tumors compared to PSMA− PC3 flu tumors.20
Here we present the synthesis and preclinical evaluation of a multimodal, PSMA-targeted dendrimer-based, theranostic agent conjugated with DM1, Cy5 NIR dye, and NOTA. The resulting PT-DDC showed preferential accumulation in PSMA-expressing tumors with efficient cellular internalization, leading to tumor growth inhibition. Despite carrying the potent cytotoxic drug DM1, the PT-DDC was well-tolerated and mice showed no signs of systemic toxicity. The Cy5 NIR dye enabled non-invasive imaging of PT-DDC accumulation in tumors during therapy and evaluation of its biodistribution, providing information regarding drug delivery and therapeutic response. However, in the case of PT-DDC, the use of Cy5 conjugation can be omitted as fluorescent dyes are primarily useful in intraoperative surgical guidance. In contrast, the use of NOTA enables labeling with radiometals such as 64Cu and 67Cu for quantitative PET imaging and radiotherapy, applicable in both pre-clinical and clinical settings.23,24 The PT-DDC, with its ability to deliver DM1 and radionuclides to PSMA-expressing tumors, presents a potential advancement in the field of theranostics for prostate cancer.
Materials and Methods
Materials
All chemicals were purchased from Sigma-Aldrich or Fisher Scientific except otherwise specified. Ethylenediamine core amine-terminated generation-4 poly(amidoamine) dendrimer G4(NH2)64 was acquired from Dendritech (Midland, MI). DM1 was purchased from BroaPharm (San Diego, CA). N-[[[(1S)-1-Carboxy-3-methylbutyl]amino]carbonyl]-L-glutamic acid (ZJ-43) and sulfo-cyanine5 NHS ester (Cy5-NHS) were purchased from Tocris Bioscience (Minneapolis, MN) and Lumirpobe Life science solutions (Hunt Valley, MD), respectively. Disuccinimidyl suberate (DSS) was acquired from TCI America (Montgomeryville, PA). L-Glutamic acid di-tert-butyl ester hydrochloride, Nε-Z-L-lysine tert-butyl ester hydrochloride and Nε-Boc-L-lysine were purchased from Chem-Impex International, Inc (Wood Dale, IL). 2,2’-(7-(2-((2,5-Dioxopyrrolidin-1-yl)oxy)-2-oxoethyl)-1,4,7-triazonane-1,4-diyl)diacetic acid (NOTA-NHS) was purchased from CD Bioparticles (Shirley, NY). 64CuCl2 (t1/2 = 12.7 h) was obtained from Washington University (St. Louis, MO). All solvents and reagents were used as received.
Synthesis
NHS-Ester-Suberic Acid Lysine-Glutamate-Urea
The NHS-SA-KEU PSMA-targeting moiety for conjugation with dendrimer was synthesized and characterized as described earlier (Figure 1A).20
 |
Figure 1 Synthesis of PSMA-targeted dendrimer-drug conjugate. (A) Synthesis of PSMA-targeting moiety terminating in amine reactive HNS ester; (B) Modification of DM1 with linker containing cleavable disulfide bond and NHS ester for conjugation with dendrimer; (C) Formulation of PSMA-targeted PT-DDC by consecutive conjugation of DM1 with cleavable linker (dashed arrow), NOTA, sulfo-Cy5, SA-KEU and capping the remaining primary amine with butane-1,2-diol groups. |
Modification of DM1
DM1 was reacted with N-Succinimidyl 3-(2-pyridyldithio)propionate (SPDP) the heterobifunctional liker to provide NHS for conjugation and dendrimer and disulfide bond for drug release (Figure 1B). DM1 (50 mg) and SPDP (25.6 mg, 1.2 mole equivalent) were dissolved in 1 mL of tetrahydrofuran (THF) and stirred under nitrogen for 24 h at room temperature (RT). Then solvent was evaporated on a rotary evaporator and the residue was reconstituted in 0.1% TFA aqueous solution with a small amount of acetonitrile (ACN) for purification on a Biotage Isolera One flash chromatography system (Salem, NH) equipped with SNAP Ultra C18 column (12 g). Gradient elution started with 100% 0.1% TFA aqueous solution and reached 100% of 0.1% TFA ACN solution in 10 column volumes. The detection wavelength was set to 254 nm. The obtained DM1-SS-NHS ester was analyzed using an Agilent 1260 liquid chromatography mass spectrometry (LC-MS) system equipped with a 6120 quadrupole spectrometer and a Luna 10 µm C18 50 × 4.6 mm column. Gradient elution starting with 10% of 0.1% TFA aqueous ACN, reaching 90% of 0.1% TFA aqueous ACN in 7 min with a flow rate at 0.7 mL/min was used (Figure S1). Chemical formula: C42H55N4O14S2, MW = 938.28 observed m/z: 938.95 (M1+1)+1, 920,48 (M1+1)+ - H2O.
Synthesis of PT-DDC and Ctrl-DDC
PT-DDC was synthesized via consecutive conjugation of DM1, NOTA, Cy5 and SA-KEU with G4(NH2)64 dendrimer and capping remaining free primary amines with butane-1,2-diol moieties, as presented in Figure 1C. G4(NH2)64 (60 mg) was dissolved in 3 mL of DMSO, and 15.8 mg (4 mole equivalent) of DM1-SS-NHS dissolved in 0.1 mL of DMSO was added. After 3 h of stirring at RT under nitrogen, conjugation of DM1 with dendrimer was confirmed by matrix-assisted laser desorption ionization-time-of-flight mass spectrometry (MALDI-TOF MS, Figure S2A), as described earlier.19,20 The resulting G4(NH2)61.7(DM1)2.3 conjugate was purified on reverse-phase high-performance liquid chromatography (RP-HPLC) to remove unconjugated DM1, as described below and lyophilized. Next, 50 mg of G4(NH2)61.7(DM1)2.3 conjugate was dissolved in 2 mL of 0.1 M NaHCO3, followed by the addition of NOTA-NHS ester (9.3 mg), sulfo-Cy5 NHS ester (16.8 mg), and NHS-SA-KEU (32.2 mg) dissolved in 0.1 mL of DMSO in 2 h intervals. Final capping of the remaining terminal primary amines was carried out overnight using 0.1 mL glycidol. Prior to addition of each reagent to the reaction mixture, 5 µL was withdrawn for MALDI-TOF MS analysis to confirm conjugation of each functional moiety, and the average number of each conjugated with dendrimer was calculated (Figure S2A). PT-DDC was purified by ultrafiltration using Amicon centrifugal filters with 10 kDa MWCO (Millipore Sigma, Rockville MD) and deionized water, followed by sterile filtration and lyophilization. PT-DDC was characterized using dynamic light scattering (DLS, Figure S2B) and RP-HPLC (Figure S2C). After conjugation of DM1, NOTA and sulfo-Cy5 with dendrimer, 0.4 mL of the reaction mixture was withdrawn and reacted with 0.05 mL of glycidol to generate non-targeted, control dendrimer-drug conjugate (Ctrl-DDC, Figure S3A), which was characterized using MALDI-TOF (Figure S3B) and DLS (Figure S3C).
MALDI-TOF MS
MALDI-TOF MS spectra were recorded on a Voyager DE-STR spectrometer, with 2,5-dihydroxybenzoic acid (DHB) as a matrix. The matrix was formulated in 50% MeOH and 0.1% TFA-H2O at a concentration of 20 mg/mL. Samples collected from the reaction mixture were purified using ZebaTM spin desalting columns with 7000 Da MWCO (ThermoFisher Scientific, Waltham MA) and diluted to 5 mg/mL with 50% MeOH and 0.1% TFA-H2O. Then 10 µL of matrix was mixed with 10 µL of sample and 1 µL of the resulting mixture was applied to the target plate. After crystallization of the matrix, the plate was used for analysis.
DLS
Dynamic light scattering and zeta potential DLS and zeta potential measurements were carried out on a Malvern Zetasizer Nano ZEN3600. PT-DDC and Ctl-DDC were prepared in PBS (c = 0.1 M, pH 7.4) at a concentration of 4 mg/mL.
RP-HPLC
The RP-HPLC system was equipped with Varian ProStar pumps, an Agilent Technology 1260 Infinity photodiode array detector and a semi-preparative C-18 Luna column (5 mm, 10×250 mm Phenomenex). Gradient elution starting with 98% H2O (0.1% TFA) and 2% ACN (0.1% TFA) reaching 90% of ACN (0.1% TFA) in 30 min at a flow rate of 2 mL/min was used. PT-DDC eluted between 3.51 and 9.65 min.
Cell Lines and Tumor Models
PSMA+ PC3 PIP and PSMA− PC3 flu cell lines were provided by Dr. Warren Heston (Cleveland Clinic, OH) and were cultured as previously described.25 The cells were authenticated by Short Tandem Repeat (STR) profile. Cells were grown to 80–90% confluency, trypsinized, and formulated in Hank’s balanced salt solution (HBSS, Sigma) for inoculation into mice. Animal studies were performed according to protocols approved by the Johns Hopkins Animal Care and Use Committee (ACUC, MO21M123). Intact male NOD-SCID mice were obtained from Johns Hopkins University in-house colony. Mice were inoculated subcutaneously in the left and right upper flanks with 1×106 PSMA+ PC3 PIP and PSMA− PC3 flu cells. Mice were used for imaging and therapy studies when the tumors reached 2.5 to 5 mm in diameter.
In vitro Binding
The in vitro PSMA specificity and affinity of PT-DDC were determined using isogenic PSMA+ PC3 PIP and PSMA− PC3 flu cell lines as described previously.19,20 Approximately 1×106 of cells were suspended in 90 µL of flow cytometry staining (FACS) buffer (1×PBS, 2 mM EDTA, 0.5% FBS), placed in FACS tubes and mixed with 10 µL of either PT-DDC or Ctrl-DDC to achieve final concentrations from 1.56×10−8 to 1×10−6 M. After 60 min incubation at RT, cells were washed with approximately 1 mL cold 1×PBS, transferred to a 96-well plate and fluorescence intensity with excitation of 645 nm and emission of 690 nm was measured using a Synergy™ 2 Multi-Mode Microplate Reader (Winooski, VT). To derive dissociation constant (Kd), the obtained binding curves were fitted to a one-site binding hyperbola using GraphPad Prism 8 software (GraphPad Software, Inc., San Diego, CA). To verify binding specificity, cells were mixed with commercially available ZJ-43 PSMA inhibitor prior addition of PT-DDC or Ctrl-DDC. All experiments were carried out in triplicate.
In vitro Cytotoxicity
PSMA+ PC3 PIP and PSMA− PC3 flu cells were seeded in 96-well plates at a density of 1×104 per well. After 24 h of incubation, PT-DDC, Ctrl-DDC and DM1 were added into cells at concentrations ranging from 1×10−5 to 1×10−12 M, in triplicate. Cells were then cultured for 48 h, followed by imaging and cell viability analysis. To evaluate uptake of PT-DDC and Ctrl-DDC by both cell types, cells were washed with growing media, and bright field and fluorescence images with excitation of 629–644 nm and emission of 661–800 nm were captured using a SparkCyto Tecan plate reader (Tecan US, Inc. Morrisville NC, Figure S4). For evaluation of cell viability, we used WST-1 assay (Abcam, Branford, CT) according to the manufacturer’s protocol. After imaging, cells were incubated with the WST-1 reagent (1:10) for 1 h. After that incubation period, the formazan dye formed was quantitated on a scanning multi-well a SparkCyto Tecan plate reader. The absorbance measured at 420 nm directly correlates to the number of viable cells. The absorbance at 600 nm was used as a passive reference.
Stability of PT-DDC
To evaluate release of DM1, 2 mg of PT-DDC was dissolved in 1 mL of 1×PBS or human blood plasma and incubated for 24 h at 37°C with gentle mixing. After incubation, 0.5 mL of the same samples were mixed with 1 mg of glutathione followed by an additional 5 h of incubation at 37°C. We used a modified capture-column method enabling direct injection of blood plasma on an HPLC system and inline solid-phase extraction of hydrophobic compounds to determine the stability of PT-DDC (Figure S5).26 An Agilent Technologies HPLC system (Santa Clara, CA) containing a 1260 Infinity quaternary pump, column compartment module and UV detector, controlled by OpenLab CDS EZChrom (A.01.04) software was used. For detection of DM1, a wavelength of 256 nm was used. The RP-HPLC system contained a manual Rheodyne injector, inline capture column packed with Phenomenex Strata-X 33 µm polymeric sorbent, and an analytical Waters 4.6×250 mm, 5 µm XBridge column. Samples loaded onto the injector were directed to the capture column and detectors with a mobile phase composed of deionized water and 0.2% of TFA at 2 mL/min. After 2 min, gradient elution starting with 100% H2O/0.2% of TFA and reaching 100% of ACN/0.2% TFA with flow rate 2 mL/min was applied to elute DM1 from the capture column to the analytical column and detectors. DM1 eluted at 17.87 min (Figure S5A). The HPLC system was calibrated using DM1 before analysis of PT-DDC incubated in 1×PBS or human blood plasma (Figure S5B). PT-DDC did not retain on the capture column and eluted within 2 min of initial isocratic elution (Figure S5C and D).
Animal Models
Animal studies were carried out according to protocols approved by the Johns Hopkins University (JHU) Animal Care and Use Committee (ACUC). For PET/CT imaging, biodistribution and therapy experiments, six-to-eight-week-old, male NOD-SCID mice were obtained from the JHU Immune Compromised Animal Core. Mice were inoculated subcutaneously in the upper flanks with 1×106 PSMA+ PC3 PIP and PSMA− PC3 flu cells and were used for experiments when tumor volume reached approximately 100 mm3.
Radiolabeling
Radiolabeling of PT-DDC and Ctrl-DDC with [64Cu] (t1/2 = 12.7 h) was carried out as we have previously described, with some modifications.19 For radiolabeling 50 µg of PT-DDC or Ctrl-DDC was dissolved in 0.1 M sodium acetate (pH 4.5) and mixed with 555 MBq (15 mCi) of [64CuCl2] and incubated of 60 min at 85°C. The resulting [64Cu]PT-DDC and [64Cu]Ctrl-DDC radiotracers were purified on ZebaTM spin desalting columns with 7000 Da MWCO (ThermoFisher Scientific, Waltham MA) pre-equilibrated with 1×PBS. For further studies, [64Cu]PT-DDC and [64Cu]Ctrl-DDC were diluted with saline and specific radioactivity was adjusted with non-radioactive PT-DDC and Ctrl-DDC.
PET/CT Imaging
Male NOD-SCID mice bearing subcutaneous PSMA+ PC3 PIP and PSMA− PC3 flu tumors were intravenously administered with approximately 10 µg of PT-DDC and 22.2 MBq (600 µCi) of [64Cu]PT-DDC formulated in 200 μL of sterile 1×PBS (n = 3). Mice were anesthetized under 2% isoflurane in oxygen at a flow rate of 2 L/min. For the blocking experiment, ZJ-43 (25 mg/kg) was co-administered with [64Cu]PT-DDC (n = 2). PET images were collected at 1, 24, 48 and 72 h after injection (ai) of the radiotracer in two bed positions to capture whole-body images (10 min per bed) on an ARGUS small-animal PET/CT scanner (Sedecal, Madrid, Spain). A computed tomography (CT) scan with 512 projections was obtained prior to each PET scan for anatomic co-registration. PET images were reconstructed using the two-dimensional ordered subsets-expectation maximization algorithm (2D-OSEM) and corrected for dead time and radioactive decay. Presented images were generated using Amira software 6.0.1 (FEI, Hillsboro, OR). Quantification of [64Cu]PT-DDC accumulation of PSMA+ PC3 PIP and PSMA− PC3 flu tumors was carried out using PMOD 4.3 (PMOD Technologies LLC).
Biodistribution of [64Cu]PT-DDC and [64Cu]Ctrl-DDC
Biodistribution studies were carried out in male NOD-SCID mice bearing PSMA+ PC3 PIP and PSMA− PC3 flu xenografts as described earlier.19,27 Mice were intravenously injected with approximately 2.22 MBq (60 µCi) of [64Cu]PT-DDC or [64Cu]Ctrl-DDC (both containing 1 µg of non-radioactive dendrimer) formulated in 200 μL of sterile 1×PBS (n = 5) and euthanized at 3, 24, 48, 72 h ai for dissection. Obtained blood and tissue samples were weighed and the radioactivity was measured using a PerkinElmer-2480 Automatic Gamma Counter (PerkinElmer, Waltham, MA). To calculate the percentage of injected dose per gram of tissue (%ID/g), triplicate standards of the radiotracers (10% of the administered dose) were counted with tissue samples. Biodistribution data shown are mean ± the standard deviation of the mean.
Optical Imaging
Biodistribution studies of PT-DDC were done in male NOD-SCID mice bearing PSMA+ PC3 PIP and PSMA− PC3 flu xenografts. Mice were intravenously injected with PT-DDC at a dose of 5 mg/kg (n = 3) and were sacrificed at 3, 24, 48 and 72 ai, followed by dissection. Dissected tissues (heart, liver, lung, pancreas, spleen, fat, kidneys, muscle, small intestines, salivary glands, bladder, bone, lacrimal gland, PSMA+ PC3 PIP tumor and PSMA− PC3 flu tumor) were imaged on a Xenogen IVIS Spectrum optical imaging system (Caliper Life Sciences, Hanover, MD) with excitation and emission set to 605 and 665 nm, respectively, and a 3 s exposure. Images were collected and processed using Living Image Software v4.5. To quantify fluorescence signal intensity of the imaged tissues, regions of interest (ROIs) were drawn around samples. Mean signal intensity in each ROI was normalized by subtracting the background signal (ROI outside tissues). Subsequently, PSMA+ PC3 PIP tumors were evaluated by epifluorescence microscopy.
Microscopy
Tumor xenografts were harvested and immediately frozen over dry ice and stored at −80°C until sectioning to a thickness of 20 μm using a Microm® HM 550 cryotome (Thermo Scientific, Waldorf, Germany), and adherence to charged glass slides (VWR, Radnor, PA). Sections were subsequently probed with anti-PSMA-FITC and anti-CD31-PE, and then scanned using the 700, 645, 550 and 345 nm channels with a Nikon 80i epifluorescence microscope. Images were displayed and analyzed using the manufacturer’s software. Fluorescent anti-PSMA (Abcam, Cambridge, UK, ab66912) and anti-CD31-PE (Novus Biologicals, Centennial, CO, NB100-1642) were labeled as follows: 100 µg of anti-PSMA was dissolved in 100 µL of PBS, pH 7.4 in a microcentrifuge tube. 1 µL of a stock solution of FITC (50 mg/mL in DMSO, LI-COR Biosciences, Lincoln, NE) was then added to the antibody solution and the labeling reaction proceeded at ambient temperature for 12 min. Unincorporated dye was removed using Sephadex G-25 size exclusion columns.
Efficacy
Mice bearing PSMA+ PC3 PIP or PSMA− PC3 flu tumors were randomized when the tumor volume reached 70 ± 40 mm3 and were subsequently treated with 2.5, 5 or 10 mg/kg of PT-DDC (n = 5) or saline as control on days 1 and 11. Tumor length and width were measured using calipers three times weekly, and tumor volumes were calculated using (L×W2)π/6, where L is the longest diameter (the major axis) and W is the tumor width, measured perpendicular to the major axis. When tumor volumes reached approximately 1000 mm3 (1083 ± 388 mm3) mice were euthanized. Mice were observed and weighed throughout the study to monitor for toxicity. Upon initiation of therapy, mice were shaved around the tumor, and imaged on a Xenogen IVIS Spectrum optical imaging system (Caliper Life Sciences, Hanover, MD) with excitation and emission at 605 and 660 nm, respectively. After measuring tumor L and W, mice were placed under 2% isoflurane in oxygen with a flow rate of 2 L/min, and images were collected to monitor for the presence of PT-DDC in tumors. Images were collected and processed using Living Image Software v4.5. To generate curves demonstrating fluorescence intensity vs time, ROIs were drawn around tumors and the mean signal intensity was derived. After euthanizing, mice were dissected to evaluate the biodistribution of PT-DDC and similar analysis was carried out to quantify signal intensity in obtained tissues, and liver, kidney and tumor samples were stored for H&E staining.
Concurrent PSMA-Targeted Delivery of DM1 and Radionuclide
To test potential of PT-DDC for simultaneous PSMA-targeted chemo- and radiotherapy male NOD-SCID mice bearing subcutaneous PSMA+ PC3 PIP and PSMA− PC3 flu tumors were intravenously administered with approximately 0.1 mg of PT-DDC (5 mg/kg) and 22.2 MBq (600 µCi) of [64Cu]PT-DDC formulated in 200 μL of PBS (n = 3). They were subsequently imaged on the ARGUS PET/CT system at 3, 24, 48 and 72 h, as described above. Upon completion of PET/CT imaging, mice were euthanized and dissected for optical imaging to evaluate the biodistribution of PT-DDC, as described above. Dissected tissues were then weighed and analyzed on a PerkinElmer-2480 Automatic Gamma Counter (PerkinElmer, Waltham, MA) to evaluate the biodistribution of [64Cu]PT-DDC.
Data Analysis
To compare and statistically analyze the treatment groups, unpaired two-tailed t-test was used, using Prism 8.2.1. Software (GraphPad). The threshold for statistical significance was set at P < 0.05.
Results
Synthesis and Physicochemical Characterization
We synthesized PT-DDC conjugated with on average 2.3 DM1 molecules, 2.9 NOTA chelators, 1.9 Cy5 NIR dyes and 20.5 SA-KEU PSMA ligands and the remaining primary amines were capped with butane-1,2-diol. NHS-SA-KEU was synthesized as described earlier (Figure 1A).20 Prior to conjugation with dendrimer, DM1 was reacted with SPDP heterobifunctional linker to provide an in vivo, cleavable disulfide bond and primary amine reactive NHS ester (Figure 1B). The purity of the resulting prodrug was confirmed by LC-MS (Figure S1). PT-DDC was formulated by consecutive conjugation of DM1, NOTA, Cy5, SA-KEU using their NHS ester-activated derivatives, followed by reacting of remaining amines with glycidol (Figure 1C). All synthetic steps were confirmed by MALDI-TOF MS (Figure S2A). Non-targeted control Ctrl-DDC was prepared using dendrimer conjugated with DM1, NOTA, Cy5 that was reacted with glycidol, omitting the conjugation of SA-KEU (Figure S3A). We derived the average number of conjugated functionalities with dendrimer by using the increase in molecular weight for the highest signal intensity in MALDI-TOF spectra (Figures S2A and S3B). DLS analysis indicated that the size distribution of PT-DDC and Ctrl-DDC averaged around 5.3 ± 1.8 and 5.1 ± 1.6 nm, respectively (Figures S2B and S3C). The zeta potentials for PT-DDC and Ctrl-DDC were −18.5 ± 2.5 and −4.5 ± 1.3 mV, respectively, indicating that they are negatively charged. RP-HPLC of PT-DDC provided a broad single peak indicating a wide range of species present in the sample. UV-vis spectrum recorded under that peak and similar profile of the chromatograms recorded using 250 and 635 nm confirmed covalent attachment of Cy5 dendrimer (Figure S2C).
In vitro Specificity of PT-DDC
PT-DDC showed high uptake in PSMA+ PC3 PIP cells that could be blocked by commercially available ZJ-43 low-molecular-weight potent PSMA inhibitor (Figure 2A), providing evidence for its PSMA specificity. In PSMA− PC3 flu cells incubated with PT-DDC, background fluorescence in the absence and presence of ZJ-43 was detected. Ctrl-DDC did not accumulate in either cell line (Figure 2A). PT-DDC also showed concentration-dependent accumulation in PSMA+ PC3 PIP cells with a Kd value of 203 ± 25 nM (Figure 2B). Specific accumulation of PT-DDC in PSMA+ PC3 PIP cells was also confirmed by imaging of wells in the plate after washing cells for cytotoxicity evaluation (Figure S4). PT-DDC demonstrated EC50 values of 4.6 nM (95% confidence interval (CI) 2.5–128.4 nM) and 89.9 nM (95% CI 43.5–185.8 nM) for PSMA+ PC3 PIP and PSMA− PC3 flu cells, respectively, further indicating selectivity for PSMA-expressing cells (Figure 2C and D). Ctrl-DDC provided a similar EC50 value of 84.6 nM (95% CI 37.7–189.7 nM) in PSMA− PC3 flu cells and higher (338.8 nM, 95% CI 59.5–1924 nM) in PSMA+ PC3 PIP cells compared to PT-DDC, indicating the need for PSMA-mediated internalization and intracellular cleavage of disulfide bonds to release DM1 for potency. Unmodified DM1 proved potent in both cell lines with EC50 values of 176.5 pM (95% CI 87.6–355.4 pM) and 89.1 pM (95% CI 55.5–995.5 pM) in PSMA+ PC3 PIP and PSMA− PC3 flu cells, respectively. The stability of PT-DDC was evaluated using RP-HPLC enabling inline solid phase extraction of DM1 on a capture column, that was subsequently eluted in to an analytical column and provided peak at 18 min (Figure S5A).26 In the chromatogram of PT-DDC incubated in 1×PBS buffer for 24 h at 37°C, only one peak associated with the conjugate was detected between 0.3 and 1.2 min as it did not retain on the capture column due to its hydrophilicity (Figure S5C). Addition of glutathione (GSH) resulted in the release of DM1 and decrease of the signal related to PT-DDC (Figure S5D). PT-DDC was also stable in human blood plasma and required addition of GSH for drug release (Figure S5E and F). PT-DDC released 2 mole equivalents of DM1 confirming conjugation on average 2 drug molecules with dendrimer.
 |
Figure 2 In vitro evaluation. (A) In vitro binding of PT-DDC and Ctrl-DDC to PSMA+ PC3 PIP and PSMA− PC3 flu cells (****P<0.001); (B) Assessment of PT-DDC affinity to PSMA; (C and D) In vitro potency of PT-DDC, Ctrl-DD and DM1 in PSMA+ PC3 PIP and PSMA− PC3 flu cells. |
PET/CT Imaging and Biodistribution
To evaluate in vivo specificity of PT-DDC we have used NOD-SCID mice bearing PSMA+ PC3 PIP and PSMA− PC3 flu tumors (Figure 3A). PET/CT imaging carried out over 3 days indicated preferential uptake of [64Cu]PT-DDC in PSMA+ PC3 PIP tumors compared PSMA− PC3 flu tumors (Figure 3A). At 3 h after injection [64Cu]PT-DDC showed the highest accumulation in bladder and kidneys, followed by liver, spleen, and heart. After radioactivity clearance from circulation, kidneys, and bladder clear delineation of PSMA+ PC3 PIP tumors was observed. In agreement with in vitro studies, co-administration of ZJ-43 with [64Cu]PT-DDC resulted in significantly lower radiotracer uptake in PSMA+ PC3 PIP tumors, confirming in vivo PSMA-specificity of PT-DDC (Figure S6A). The uptake of the radioactivity in peripheral organs was similar in mice with and without blocking. Correlative biodistribution results showed a consistently high accumulation of [64Cu]PT-DDC with a percent of injected per gram (%ID/g) of 14.3 ± 0.2, 11.7 ± 0.5, 10.8 ± 1.6 and 13.2 ± 0.5 in PSMA+ PC3 PIP tumors at 3, 24, 48 and 72 h ai, respectively (Figure 3B, Table S1). In PSMA− PC3 flu tumors, the %ID/g values were significantly lower 2.4 ± 0.2, 3.9 ± 0.5, 3.6 ± 0.5 and 3.1 ± 0.5 at the same time points, indicating higher radioactivity accumulation in PSMA+ PC3 PIP tumors. Highest PSMA+ PC3 PIP tumor-to-blood of 8.3 ± 0.6 and PSMA+ PC3 PIP-to-muscle of 34.3 ± 9.8 ratios were observed at 72 h ai. In liver the %ID/g was 13.7 ± 0.9, 16.1 ± 2.3, 12.5 ± 1.6 and 11.12 ± 0.6 at 3, 24, 48 and 72 h ai, respectively. In spleen the radioactivity accumulation was 6.7 ± 1.5, 8.6 ± 1.7, 6.8 ± 0.7 and 5.1 ± 1.3 %ID/g at the same time points. In agreement with renal clearance of [64Cu]PT-DDC the radioactivity accumulation in kidneys was 37.5 ± 2.7, 15.8 ± 2.7, 10.2 ± 1.1 and 9.3 ± 0.4 %ID/g at 3, 24, 48 and 72 h ai. The radioactivity accumulation in other tissues analyzed was below 5 %ID/g. PSMA-specificity of PT-DDC was further confirmed by biodistribution of [64Cu]Ctrl-DDC that showed low and similar accumulation in PSMA+ PC3 PIP and PSMA− PC3 flu tumors with %ID/g of 2.4 ± 0.1 and 2.2 ± 0.1, respectively at 3 h ai (Figure S6B and Table S1). The highest uptake of [64Cu]Ctrl-DDC was detected in kidneys (42.2 ± 2.5 %ID/g) followed by liver (8.9 ± 0.5 %ID/g), confirming major renal clearance. In vivo specificity of PT-DDC was also confirmed using optical imaging (Figure 3C). The highest fluorescence intensity was detected in kidneys followed by PSMA+ PC3 PIP tumors and the lowest in PSMA− PC3 flu tumors, muscles, fat, spleen and heart at 3 h ai. After clearance of PT-DDC from blood vessels, the fluorescence intensity significantly decreased in all off-target tissues and remained relatively high in PSMA+ PC3 PIP tumors. The PSMA+ PC3 PIP/PSMA− PC3 flu tumors ratio of 9.6 ± 2.4, 11.4 ± 0.8, 10.7 ± 1.6 and 10.1 ± 1.4 at 3, 24, 48 and 72 h ai, respectively, indicated approximately 10-fold higher accumulation of PT-DDC in PSMA+ PC3 PIP tumors compared with PSMA− PC3 flu tumors and other off-target tissues except kidneys. PSMA+ PC3 PIP/kidneys ratios were 0.7 ± 0.1, 2.2 ± 0.6, 2.7 ± 0.9 and 3.1 ± 0.5 at 3, 24, 48 and 72 h ai, respectively, consistent with renal clearance of PT-DDC. Microscopy results collected for PSMA+ PC3 PIP tumors, obtained from these mice, showed very low accumulation of PT-DDC in peritumoral fat and its high presence in the tumoral edge at 3 h ai (Figure 3D). The uptake of PT-DDC in tumor parenchyma was associated with tumor vasculature and it accumulated in cells nearby. Areas with low vascular density display little PT-DDC uptake (Figure 3D image iii upper right quadrant).
 |
Figure 3 Biodistribution. (A) Representative decay-corrected PET/CT volume-rendered images of mice bearing PSMA+ PC3 PIP (grey arrows) and PSMA− PC3 flu (white arrows) tumors injected with of 22.2 MBq (600 µCi) of [64Cu]PT-DDC (n=3, ai - after injection, D - dorsal, V–ventral); (B) Biodistribution of [64Cu]PT-DDC in the same PC model (n=5, mice were injected with 60 µCi, **** P<0.001); (C) Representative optical images of tissues dissected from mice bearing PSMA+ PC3 PIP and PSMA− PC3 flu tumors injected with 5 mg/kg (~100 ug) of PT-DDC and its semi-quantitative biodistribution (n=3, numbers above bars reflect numbered tissues); (D) Distribution of PT-DDC in PSMA+ PC3 PIP tumors at microscopic level (i) peritumoral fat: white - PT-DDC, green - tissue autofluorescence, red - anti-CD31-PE(R), blue - hoechst33432 (3 h ai, scale: 320.09 µm x 320.09 µm), (ii) tumor edge: white - PT-DDC, green - anti-PSMA-FITC, blue - hoechst33432 (scale: 320.09 µm x 320.09 µm), (iii) tumor parenchyma: white - PT-DDC, red - anti-CD31-PE(R), blue - hoechst33432 (scale: 1280.35 µm x 1280.35 µm), (iv) tumor parenchyma showing PT-DDC deposition associated with vasculature: white - PT-DDC, green - anti-PSMA-FITC, red - anti-CD31-PE(R), blue - hoechst33432. |
Efficacy
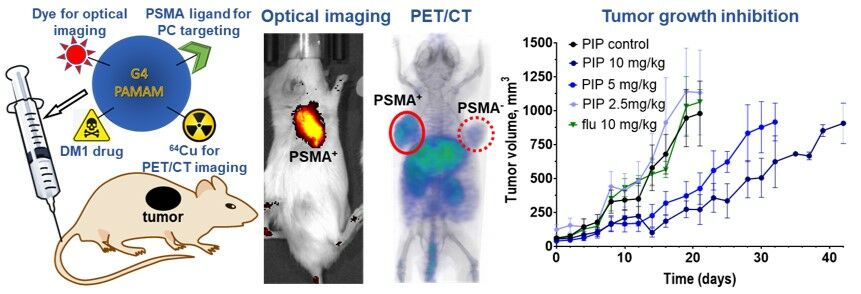
The evaluation of PT-DDC therapeutic efficacy is presented in Figure 4. Three weeks after inoculation the average tumor volume reached 70 ± 45 mm3, and mice were IV injected with 2.5, 5 and 10 mg/kg of PT-DDC on day 1 and day 11. Mice did not show any signs of toxicity and maintained weight throughout the entire experiment (Figure S7) and were sacrificed when the tumor volume reached approximately 1000 mm3. PSMA+ PC3 PIP tumors in control and treatment (2.5 mg/kg) groups as well as PSMA− PC3 flu tumors in mice treated with 10 mg/kg of PT-DDC, grew relatively fast and those animals were euthanized on day 21 (Figure 4A). Relative tumor volumes indicated that PT-DDC showed dose dependent tumor growth inhibition (TGI) in mice bearing PSMSA+ PC3 PIP tumors, especially after the second administration of PT-DDC (Figure 4B). The %TGI was 82.6 ± 8.2, 46.9 ± 21.4 and 43.3 ± 10.8 for 10, 5 and 2.5 mg/kg dose, respectively on day 14 (Figure 4C). The %TGI was 73.4 ± 13.4, 53.4 ± 22.1 and 34.7 ± 10.3 for the same doses on day 21 (Figure 4C). Treatment of mice bearing PSMSA+ PC3 PIP with PT-DDC at 5, and 10 mg/kg dose prolonged survival to 32 and 42 days, respectively. The presence of PT-DDC in tumors was monitored using non-invasive optical imaging (Figure 4D and E). In agreement with biodistribution results, accumulation of PT-DDC was detected in PSMSA+ PC3 PIP tumors but it did not provide detectable signal in PSMA− PC3 flu tumors (Figure 4D). The second dose of PT-DDC was administered on day 11 after a substantial decrease of fluorescence intensity in PSMSA+ PC3 PIP tumors was detected (Figure 4D and E). The second administration of PT-DDC resulted in a significant increase of fluorescence intensity in targeted tumors (Figure 4D and E), followed by enhanced TGI. In contrast, administration of 10 mg/kg PT-DDC in mice bearing PSMSA− PC3 flu xenografts resulted in significantly lower increase of florescence intensity in tumors and no TGI. Biodistribution of PT-DDC in mice bearing PSMA+ PC3 PIP tumors, obtained 10 days after the second 2.5 mg/kg dose, indicated its selective and prolonged retention in targeted tumors. The H&E staining carried out using tissues obtained from mice treated with 10 mg/kg PT-DDC did not show toxicity in kidneys and liver and high necrosis was detected in PSMA+ PC3 PIP tumors, while no necrosis was detected in PSMA− PC3 flu tumors obtained from mice treated with the same dose (Figure 4G).
 |
Figure 4 Therapy study. Mice bearing PSMSA+ PC3 PIP or PSMSA− PC3 flu were administered 2.5, 5 and 10 mg/kg of PT-DDC on day 1 and 11 (n=5). (A) Tumor volumes (arrow indicates second administration of PT-DDC); (B) relative tumor volume; (C) Tumor growth inhibition on day 14 (i) and 21 (ii); (D) Representative optical imaging of mice bearing PSMSA+ PC3 PIP treated with 10 mg/kg of PT-DDC (d - day, ai - after injection); (E) Fluorescence intensity detected within tumors in all treatment groups; (F) Biodistribution of PT-DDC 10 days after injection of the 2nd dose of 2.5 mg/kg on day 11 of the treatment in mice bearing PSMA+ PC3 PIP tumors; (G) Representative H&E staining of kidney, liver, PSMA+ PC3 PIP and PSMA− PC3 flu tumors obtained from mice injected with 10 mg/kg of PT-DDC. Results indicate dose dependent accumulation of PT-DDC in PSMA+ PC3 PIP tumors and therapeutic response and its prolonged retention in these tumors. |
Concurrent PSMA-Specific Drug and Radionuclide Delivery
Imaging studies of mice bearing PSMA+ PC3 PIP and PSMA− PC3 flu injected with a therapeutic dose of PT-DDC (5 mg/kg) and 22.2 MBq (600 µCi) of [64Cu]PT-DDC indicated potential for simultaneous PC-targeted chemo- and radiotherapy (Figure 5). The radioactivity accumulation in PSMA+ PC3 PIP tumors was 8.5 ± 1.5, 12.2 ± 2.3, 14.2 ± 1.8 and 13.5 ± 2.5 %ID/cc at 3, 24, 48 and 72 h ai. The radioactivity accumulation of PSMA− PC3 flu tumors was below 4 %ID/cc at the same time points. Consistent with above-described results PET/CT imaging showed the presence of radioactivity in bladder, kidneys, liver, lungs, heart, and the periphery of PSMA+ PC3 PIP tumors at 3 h ai. At later time points the radioactivity remained in PSMA+ PC3 PIP tumors, liver, spleen and kidneys. Upon completion of PET/CT imaging mice were dissected and tissues were analyzed using optical imaging showing the highest fluorescence intensity in PSMA+ PC3 PIP tumors followed by kidneys (Figure 5B). The PSMA+ PC3 PIP/PSMA− PC3 flu tumors ratio was 9.2 ± 3.1 and PSMA+ PC3 PIP tumors/kidneys ratio was 3.4 ± 0.9 indicating significantly higher PT-DDC accumulation in PSMA+ PC3 PIP tumors compared to off-target tissues, in agreement with initial biodistribution evaluation of PT-DDC presented in the Figure 3C and described above. Then dissected tissues were analyzed in a gamma counter to obtain biodistribution of [64Cu]PT-DDC, demonstrating higher accumulation of PSMA+ PC3 PIP tumors (15.3 ± 1.1 %ID/g) vs PSMA− PC3 flu tumors (5.4 ± 0.2%ID/g). The accumulation of radioactivity in liver, kidneys and spleen was 18.2 ± 1.5, 14.1 ± 1.2 and 12.2 ± 2.8 %ID/g, respectively.
 |
Figure 5 Evaluation of concurrent delivery of DM1 and radioactivity to PSMA+ PC3 PIP tumors at therapeutic dose. (A) Representative decay-corrected PET/CT volume-rendered (i), axial (ii), coronal (iii) and sagittal (iv) images (ii, iii and iv were generated using center of PSMA+ PC3 PIP tumor and 1 mm thickness) of mice bearing PSMA+ PC3 PIP and PSMA− PC3 flu tumors injected with 5 mg/kg of PT-DDC and 22.2 MBq (600 µCi) of [64Cu]PT-DDC (n=3). After completion of PET/CT imaging mice were dissected and harvested tissues were imaged on an optical scanner and analyzed on a gamma counter to evaluate biodistribution of PT-DDC and [64Cu]PT-DDC, respectively. (B) Representative optical image of dissected tissues and semi-quantitative biodistribution of PT-DDC (numbers above bars reflect numbered tissues) and (C) biodistribution of [64Cu]PT-DDC, ****P<0.001. Results indicate feasibility of concurrent PSMA-specific chemo- and radiotherapy by PT-DDC. |
Discussion
Our focus is to implement PAMAM dendrimers to treat PC. We have demonstrated that generation-4 PAMAM dendrimers conjugated with KEU for targeting PSMA and multiple other moieties used for imaging showed preferential uptake in PSMA+ tumors vs off-target tissues.19,20 To evaluate our theranostic strategy further, we studied PSMA-targeted dendrimers conjugated with DM1, a potent antimitotic agent commonly used in ADCs, Cy5 fluorescent dye for optical imaging, and NOTA chelator for radiolabeling with 64Cu, enabling PET/CT imaging and biodistribution studies. The design of this multimodal PT-DDC allowed comprehensive in vivo characterization with non-invasive imaging, which provided a correlation between tumor-specific accumulation and therapeutic responses. Despite the extensive dendrimer surface modification with drug, imaging, chelating, targeting and capping moieties, PT-DDC showed specific uptake in PSMA+ PC3 PIP tumors through active PSMA targeting and renal clearance, facilitated by a size distribution averaging around 5 nm and negative-net surface charge. Only two intravenous injections of PT-DDC resulted in significant tumor growth inhibition at doses used for ADCs, without apparent side effects. Using PET/CT and optical imaging, we also demonstrated the potential of our PSMA-targeted dendrimers for concurrent PC-specific delivery of chemotherapeutics and radionuclides at therapeutic doses, suitable for β- or α-particle-emitting radioisotopes. In that line, a combination of external beam radiation followed by chemo- or immunotherapy are currently being used in several cancers, with enhanced therapeutic outcomes over single-modality approaches.28
Clinically evaluated PSMA-targeted radiotherapeutics, ADCs and BIND-014 nanoparticles have provided promising results, however they are not curative and have limited options for improvement in efficacy and systemic toxicity. The most common adverse effects of low-molecular-weight PSMA-targeted radiotherapeutics include thrombopenia, xerostomia and nephrotoxicity due to their high accumulation in salivary glands and kidneys.5,6,8,11 PSMA-specific ADCs showed significant peripheral neuropathy at therapeutic doses.9,14,15 In contrast, PAMAM dendrimers can be conjugated with multiple therapeutic and targeting agents, and their pharmacokinetics can be optimized by simply using different generations or changing surface functionalization to promote either active or passive cancer targeting.21,29,30 Sadekar et al demonstrated that hydroxyl-terminated PAMAM generation-5, -6 and -7 dendrimers with size distributions around 5, 6 and 7 nm, respectively, showed vastly different pharmacokinetics in mice bearing A2780 human ovarian carcinoma, despite the minimal size discrepancy.30 Generation-5 dendrimers were rapidly excreted by renal filtration with major uptake in kidneys. Generation-6 dendrimers showed the highest uptake in liver followed by kidneys and spleen, with marginal tumor uptake. Generation-7 dendrimers showed prolonged circulation and passive accumulation in tumors reaching approximately 7 %ID/g.30 We have recently demonstrated that CXCR4-targeted PAMAM dendrimers with a size distribution averaging around 8 nm showed predominantly passive cancer targeting.27 Conjugation of CXCR4-specific peptides with dendrimers resulted in faster and prolonged retention in CXCR4+ tumors, but the accumulation level was similar for targeted and control dendrimers.27 Zhu et al evaluated the therapeutic efficacy of extensively PEGylated generation-4 PAMAM dendrimer conjugated with doxorubicin and cyclic cRDG peptide, targeting αvβ3 integrins, with a size distribution of around 17 nm and a nearly neutral surface, with zeta potential of 2.31± 0.15 mV in a murine B16 melanoma model.31 While non-targeted control and αvβ3-targeted dendrimer conjugates showed improved therapeutic efficacy over doxorubicin, they promoted similar tumor growth inhibition and survival.31 Based on the above mentioned considerations, it is apparent that the size of non-targeted control and αvβ3-targeted dendrimer conjugates would facilitate mostly passive tumor targeting, which is supported by the similar biodistribution of doxorubicin upon administration of both conjugates.31 Baker’s group, who pioneered development of PAMAM dendrimer-based therapeutic agents, reported promising preclinical evaluation of G5-FA-MTX, acylated generation-5 dendrimers conjugated with folic acid (FA) and methotrexate (MTX).32,33 However, no pharmacokinetic data were reported for G5-FA-MTX, except for a report on a tritiated G5-3H-FA conjugate in the murine KB human epithelial cancer murine model, showing accumulation of up to 8 %ID/g in tumors with relatively fast clearance from circulation within 24 h ai, with accumulation primarily in liver, kidneys and spleen.32 In contrast, PT-DDC with a size distribution of around 5 nm showed predominantly active targeting and renal clearance with low off-target uptake. The accumulation of [64Cu]PT-DDC in PSMA+ PC3 PIP tumors reached nearly 14% ID/g, with a PSMA+ PC3 PIP/PSMA− PC3 flu tumors ratio of around 4. Optical imaging, however, indicated a 10-fold higher accumulation of PT-DDC in PSMA+ PC3 PIP tumors than in PSMA− PC3 flu tumors and other tissues. The observed discrepancies in biodistribution of PT-DDC obtained using optical imaging and [64Cu]PT-DDC are due mainly to 64Cu2+ transchelation, as we have demonstrated for our PSMA-targeted dendrimer conjugated with DOTA.19 While NOTA forms a thermodynamically stable complex with 64Cu2+, we still observed its transchelation, most likely owing to dendrimers weakly coordinating 64Cu2+. PAMAM dendrimers were shown to coordinate Cu2+ weakly by 2–4 dendrimer tertiary amine groups located in the equatorial plane and 2 axial water molecules.34,35 The number of NOTA chelators per dendrimer can be optimized to improve radiolabeling and minimize off-site chelation. NOTA can also be used for chelation of 67Cu2+ enabling radiotherapy, which is a β− emitter with a half-life (t1/2) of 2.58 days. For PC-specific concurrent chemo- and radiotherapy with PSMA-targeted dendrimers, conjugation of DOTA for radiolabeling with 203Pb and 212Pb (t1/2 = 10.64 h), allowing single photon emission computed tomography (SPECT) and α therapy, may be more appropriate options. DOTA can also be used for radiolabeling with alpha and beta emitting isotopes 225Ac (t1/2 = 10 days) and 177Lu (t1/2 = 6.647 days). Application of 225Ac and 177Lu with longer half-lives may be beneficial due to prolonged retention of PT-DDC in PSMA+ PC3 PIP tumors as demonstrated. PSMA-targeted dendrimer theranostics enabling concurrent chemo- and radiotherapy may be advantageous for treatment of aggressive PC for robust target engagement and minimal off-target toxicity, which is challenging with currently available PSMA-targeted low-molecular-weight radiotherapeutics or ADCs.
Conclusions
Our results demonstrate the efficacy of PSMA-targeted PAMAM dendrimer-based theranostic agents for image-guided treatment of PC. Direct quantitative imaging of drug delivery provides information needed to understand and improve therapeutic efficacy.36 PT-DDC showed preferential and prolonged accumulation in PSMA+ tumors mediated by active targeting, as demonstrated by non-invasive optical and PET/CT imaging. Accordingly, treatment of mice bearing PSMA+ tumors with PT-DDC resulted in significant tumor growth inhibition and extended survival without apparent systemic toxicity. PT-DDC also demonstrated potential for simultaneous PC-specific chemo- and radiotherapy, an avenue we are currently exploring. However, conjugation of Cy5 NIR dye can be omitted for clinical utility. This streamlines manufacturing of PT-DDC for clinical translation.
Abbreviations
PSMA, Prostate-specific membrane antigen; PT-DDC, prostate-specific membrane antigen targeted dendrimer-drug conjugate; Ctrl-DDC control dendrimer-drug conjugate; PET/CT, positron emission tomography/computed tomography, d - day, ai - after injection, %ID/g - percent of injected per gram.
Acknowledgments
We thank The Johns Hopkins University School of Medicine MRB Molecular Imaging Service Center and Cancer Functional Imaging Core for collecting PET/CT imaging data. This paper was presented at the World Molecular Imaging Congress as a conference talk with interim findings. The talk’s abstract was published in 2022 World Molecular Imaging Congress Program in Molecular Imaging and Biology: [https://link.springer.com/article/10.1007/s11307-022-01794-2].
Funding
This work was funded by CA134675, CA184228, CA183031, EB024495, US DoD CDMRP W81XWH-18-1-0188, and the Commonwealth Foundation.
Disclosure
Dr Martin Pomper and Dr Wojciech Lesniak report a patent US-20220054659-A1 and US-20220401592 issued to n/a, a patent US-20220401592-A1 and US-20220401592 issued. The authors report no other conflicts of interest in this work.
References
1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–386. doi:10.1002/ijc.29210
2. Chen RC, Rumble RB, Loblaw DA, et al. Active surveillance for the management of localized prostate cancer (Cancer Care Ontario Guideline): American Society of Clinical Oncology Clinical Practice Guideline Endorsement. J clin oncol. 2016;34(18):2182–2190. doi:10.1200/JCO.2015.65.7759
3. Damodaran S, Lang JM, Jarrard DF. Targeting metastatic hormone sensitive prostate cancer: chemohormonal therapy and new combinatorial approaches. J Urol. 2019;201(5):876–885. doi:10.1097/JU.0000000000000117
4. James ND, Spears MR, Clarke NW, et al.; S. Investigators. Failure-free survival and radiotherapy in patients with newly diagnosed nonmetastatic prostate cancer: data from patients in the control arm of the STAMPEDE trial. JAMA Oncol. 2016;2(3):348–357. doi:10.1001/jamaoncol.2015.4350
5. Kiess AP, Banerjee SR, Mease RC, et al. Prostate-specific membrane antigen as a target for cancer imaging and therapy. Q J Nucl Med Mol Im. 2015;59:241–268.
6. Maurer T, Eiber M, Schwaiger M, Gschwend JE. Current use of PSMA - PET in prostate cancer management. Nat Rev Urol. 2016;13(4):226–235. doi:10.1038/nrurol.2016.26
7. Phillips R, Shi WY, Deek M, et al. Outcomes of observation vs stereotactic ablative radiation for oligometastatic prostate cancer: the ORIOLE Phase 2 randomized clinical trial. JAMA Oncol. 2020;6(5):650. doi:10.1001/jamaoncol.2020.0147
8. Han S, Woo S, Kim YJ, Suh CH. Impact of Ga-68-PSMA PET on the management of patients with prostate cancer: a systematic review and meta-analysis. Eur Urol. 2018;74(2):179–190. doi:10.1016/j.eururo.2018.03.030
9. Petrylak DP, Kantoff P, Vogelzang NJ, et al. Phase 1 study of PSMA ADC, an antibody-drug conjugate targeting prostate-specific membrane antigen, in chemotherapy-refractory prostate cancer. Prostate. 2019;79(6):604–613. doi:10.1002/pros.23765
10. Von Hoff DD, Mita MM, Ramanathan RK, et al. Phase I study of PSMA-targeted docetaxel-containing nanoparticle BIND-014 in patients with advanced solid tumors. Clin Cancer Res. 2016;22(13):3157–3163. doi:10.1158/1078-0432.CCR-15-2548
11. Wester HJ, Schottelius M. PSMA-targeted radiopharmaceuticals for imaging and therapy. Semin Nucl Med. 2019;49(4):302–312. doi:10.1053/j.semnuclmed.2019.02.008
12. Wondergem M, van der Zant FM, Lazarenko SV, Knol RJJ. F-18-DCFPyL PET/CT in primary staging of prostate cancer. Eur J Nucl Med Mol. 2018;45:S535–S535.
13. Keam SJ. Lutetium Lu 177 vipivotide tetraxetan: first approval. Mol Diagn Ther. 2022;26(4):467–475. doi:10.1007/s40291-022-00594-2
14. Coats S, Williams M, Kebble B, et al. Antibody-drug conjugates: future directions in clinical and translational strategies to improve the therapeutic index. Clin Cancer Res. 2019;25(18):5441–5448. doi:10.1158/1078-0432.CCR-19-0272
15. Tolcher AW. Antibody drug conjugates: lessons from 20 years of clinical experience. Ann Oncol. 2016;27(12):2168–2172. doi:10.1093/annonc/mdw424
16. Mita M, Burris H, LoRusso P, et al. A phase 1 study of BIND-014, a PSMA-targeted nanoparticle containing docetaxel, administered to patients with refractory solid tumors on a weekly schedule. Cancer Res. 2014;74(19_Supplement):CT210. doi:10.1158/1538-7445.AM2014-CT210
17. Natale R, Socinski M, Hart L, et al. Clinical activity of BIND-014 (docetaxel nanoparticles for injectable suspension) as second-line therapy in patients (pts) with Stage III/IV non-small cell lung cancer. Eur J Cancer. 2014;50:19. doi:10.1016/S0959-8049(14)70167-4
18. Banerjee SR, Foss CA, Horhota A, et al. 111 In- and IRDye800CW-labeled PLA–PEG nanoparticle for imaging prostate-specific membrane antigen-expressing tissues. Biomacromolecules. 2017;18(1):201–209. doi:10.1021/acs.biomac.6b01485
19. Lesniak WG, Boinapally S, Banerjee SR, et al. Evaluation of PSMA-targeted PAMAM dendrimer nanoparticles in a murine model of prostate cancer. Mol Pharm. 2019;16(6):2590–2604. doi:10.1021/acs.molpharmaceut.9b00181
20. Lesniak WG, Wu YX, Kang J, et al. Dual contrast agents for fluorescence and photoacoustic imaging: evaluation in a murine model of prostate cancer. Nanoscale. 2021;13(20):9217–9228. doi:10.1039/D1NR00669J
21. Araujo RV, Santos SDS, Igne Ferreira E, Giarolla J. New advances in general biomedical applications of PAMAM dendrimers. Molecules. 2018;23(11):2849. doi:10.3390/molecules23112849
22. Kim Y, Park EJ, Na DH. Recent progress in dendrimer-based nanomedicine development. Arch Pharm Res. 2018;41(6):571–582. doi:10.1007/s12272-018-1008-4
23. Banerjee SR, Pullambhatla M, Foss CA, et al. 64Cu-labeled inhibitors of prostate-specific membrane antigen for PET imaging of prostate cancer. J Med Chem. 2014;57(6):2657–2669. doi:10.1021/jm401921j
24. Carlos Dos Santos J, Beijer B, Bauder-Wust U, et al. Development of novel PSMA ligands for imaging and therapy with copper isotopes. J Nucl Med. 2020;61(1):70–79. doi:10.2967/jnumed.119.229054
25. Mease RC, Dusich CL, Foss CA, et al. N-[N-[(S)-1,3-Dicarboxypropyl]Carbamoyl]-4-[18F]Fluorobenzyl-l-Cysteine, [18F]DCFBC: a new imaging probe for prostate cancer. Clin Cancer Res. 2008;14(10):3036–3043. doi:10.1158/1078-0432.CCR-07-1517
26. Hilton J, Yokoi F, Dannals RF, Rauert HT, Szabo Z, Wong DF. Column-switching HPLC for the analysis of plasma in PET imaging studies. Nucl Med Biol. 2000;27(6):627–630. doi:10.1016/S0969-8051(00)00125-6
27. Lesniak WG, Azad BB, Chatterjee S, Lisok A, Pomper MG. An evaluation of CXCR4 targeting with PAMAM dendrimer conjugates for oncologic applications. Pharmaceutics. 2022;14(3):655. doi:10.3390/pharmaceutics14030655
28. Liu YP, Zheng CC, Huang YN, He ML, Xu WW, Li B. Molecular mechanisms of chemo- and radiotherapy resistance and the potential implications for cancer treatment. Medcomm. 2021;2(3):315–340. doi:10.1002/mco2.55
29. Kannan RM, Nance E, Kannan S, Tomalia DA. Emerging concepts in dendrimer-based nanomedicine: from design principles to clinical applications. J Intern Med. 2014;276(6):579–617. doi:10.1111/joim.12280
30. Sadekar S, Ray A, Janat-Amsbury M, Peterson CM, Ghandehari H. Comparative biodistribution of PAMAM dendrimers and HPMA copolymers in ovarian-tumor-bearing mice. Biomacromolecules. 2011;12(1):88–96. doi:10.1021/bm101046d
31. Zhu SJ, Qian LL, Hong MH, Zhang LH, Pei YY, Jiang YY. RGD-modified PEG-PAMAM-DOX conjugate: in vitro and in vivo targeting to both tumor neovascular endothelial cells and tumor cells. Adv Mater. 2011;23(12):H84–H89. doi:10.1002/adma.201003944
32. Kukowska-Latallo JF, Candido KA, Cao ZY, et al. Nanoparticle targeting of anticancer drug improves therapeutic response in animal model of human epithelial cancer. Cancer Res. 2005;65(12):5317–5324. doi:10.1158/0008-5472.CAN-04-3921
33. Majoros IJ, Thomas TP, Mehta CB, Baker JR. Poly(amidoamine) dendrimer-based multifunctional engineered nanodevice for cancer therapy. J Med Chem. 2005;48(19):5892–5899. doi:10.1021/jm0401863
34. Diallo MS, Balogh L, Shafagati A, et al. Dendritic chelating agents for water treatment: experimental investigations and modeling of Cu(II) binding in aqueous solutions of poly(amidoamine) dendrimers. Abstr Pap Am Chem S. 2000;219:U654–U654.
35. Diallo MS, Christie S, Swaminathan P, et al. Dendritic chelating agents. 1. Cu(II) binding to ethylene diamine core poly(amidoamine) dendrimers in aqueous solutions. Langmuir. 2004;20(7):2640–2651. doi:10.1021/la036108k
36. Chakravarty R, Hong H, Cai W. Positron emission tomography image-guided drug delivery: current status and future perspectives. Mol Pharm. 2014;11(11):3777–3797. doi:10.1021/mp500173s
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.