")
Back to Journals » Drug Design, Development and Therapy » Volume 18
MTCH2 in Metabolic Diseases, Neurodegenerative Diseases, Cancers, Embryonic Development and Reproduction
Authors Peng X , Yang Y, Hou R, Zhang L, Shen C, Yang X, Luo Z, Yin Z , Cao Y
Received 30 January 2024
Accepted for publication 21 May 2024
Published 12 June 2024 Volume 2024:18 Pages 2203—2213
DOI https://doi.org/10.2147/DDDT.S460448
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Xiaoqing Peng,1– 4,* Yuanyuan Yang,3,4,* Ruirui Hou,1 Longbiao Zhang,1 Can Shen,3 Xiaoyan Yang,3 Zhigang Luo,5 Zongzhi Yin,3,4 Yunxia Cao3,4
1School of Pharmacy, Anhui Medical University, Hefei, People’s Republic of China; 2Inflammation and Immune Mediated Diseases Laboratory of Anhui Province, Anhui Institute of Innovative Drugs, Hefei, Anhui, People’s Republic of China; 3Department of Obstetrics and Gynecology, The First Affiliated Hospital of Anhui Medical University, Hefei, People’s Republic of China; 4The Key National Health Commission Key Laboratory of Study on Abnormal Gametes and Reproductive Tract (Anhui Medical University), Hefei, People’s Republic of China; 5Department of Cardiology, The First Affiliated Hospital of Anhui Medical University, Hefei, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yunxia Cao; Zongzhi Yin, The First Affiliated Hospital of Anhui Medical University, No. 218 Jixi Road, Sushan District, Hefei, Anhui, People’s Republic of China, Email [email protected]; [email protected]
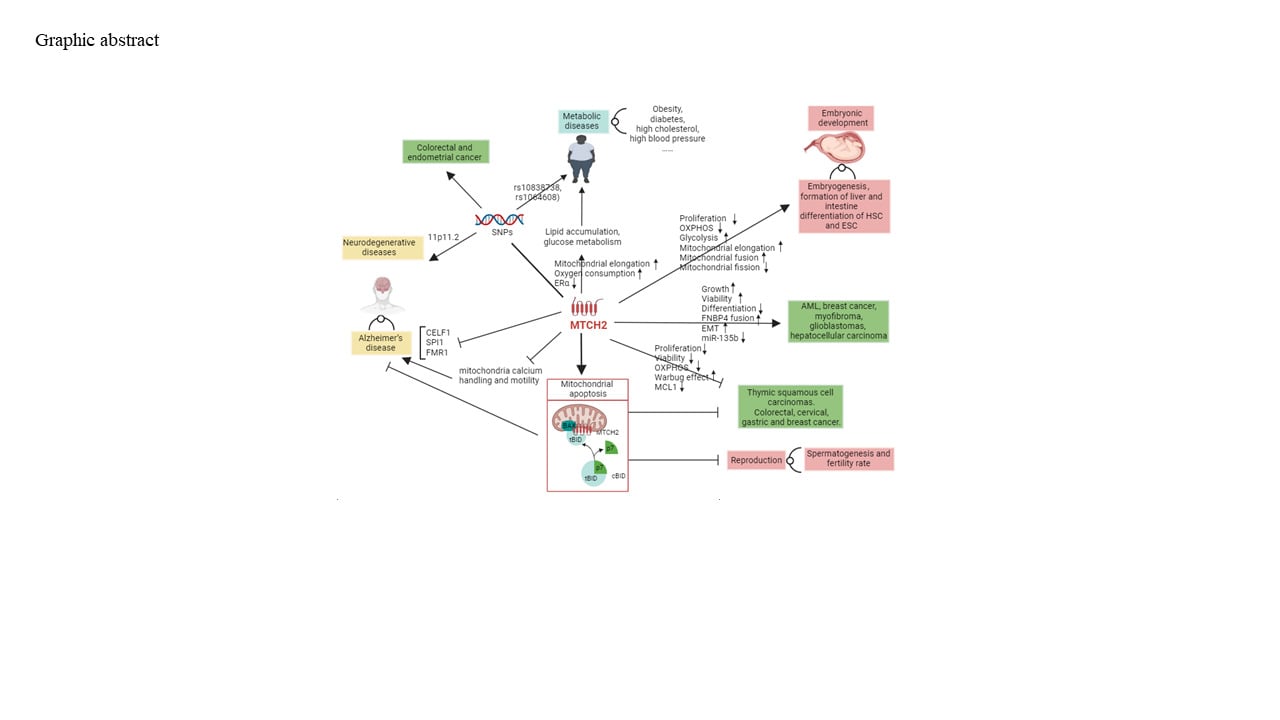
Abstract: Mitochondrial carrier homolog 2 (MTCH2) is a member of the solute carrier 25 family, located on the outer mitochondrial membrane. MTCH2 was first identified in 2000. The development in MTCH2 research is rapidly increasing. The most well-known role of MTCH2 is linking to the pro-apoptosis BID to facilitate mitochondrial apoptosis. Genetic variants in MTCH2 have been investigated for their association with metabolic and neurodegenerative diseases, however, no intervention or therapeutic suggestions were provided. Recent studies revealed the physiological and pathological function of MTCH2 in metabolic diseases, neurodegenerative diseases, cancers, embryonic development and reproduction via regulating mitochondrial apoptosis, metabolic shift between glycolysis and oxidative phosphorylation, mitochondrial fusion/fission, epithelial-mesenchymal transition, etc. This review endeavors to assess a total of 131 published articles to summarise the structure and physiological/pathological role of MTCH2, which has not previously been conducted. This review concludes that MTCH2 plays a crucial role in metabolic diseases, neurodegenerative diseases, cancers, embryonic development and reproduction, and the predominant molecular mechanism is regulation of mitochondrial function. This review gives a comprehensive state of current knowledgement on MTCH2, which will promote the therapeutic research of MTCH2.
Keywords: Mimp, MTCH2, mitochondrial carrier homolog 2, BID, apoptosis
Graphical Abstract:
Introduction
Mitochondrial carrier homolog 2 (MTCH2) is a member of the solute carrier 25 family, an integral membrane protein known for the role of catalyzing the exchange of substrates across the inner mitochondrial membrane (IMM). MTCH2 is primarily located on the outer mitochondrial membrane (OMM) rather than the IMM, and functions as an insertase1 and also a scramblase.2,3 The insertase allows the insertion of cytoplasmic α-helical protein into the outer mitochondrial membrane but not the transfer to the inner mitochondrial membrane.1 The scramblase facilitates bidirectional lipid transport across cell membranes.4 The best-known role of MTCH2 is binding with truncated BID (tBID) to regulate mitochondrial apoptosis.5 Recent studies have demonstrated the physiological and pathological role of MTCH2 in metabolic diseases, neurodegenerative diseases, cancers, embryonic development and reproduction, and the predominant molecular mechanisms are regulating apoptosis, metabolic shift, mitochondrial elongation, epithelial-mesenchymal transition, etc. No review has summarised those findings with disease-oriented aspects, although a recent review article published in March 2024 concluded current studies on MTCH2 from a mechanism of action aspects. A thoughtful understanding of MTCH2 in health and diseases is helpful in guiding future research. Therefore, a total of 130 articles related to MTCH2 are systematically searched using PubMed databases, and the findings are summarised in this review. In the following sections, we will demonstrate the structure and pro-apoptosis role of MTCH2 and summarise the current studies of MTCH2 in metabolic diseases, neurodegenerative diseases, cancers, embryonic development and reproduction.
 |
Figure 1 Structure of MTCH2. (A) 303 amino acids of human MTCH2, red refers to the position cross-linking to cBID; yellow refers to the amino acids 123 to 206, which construct a mitochondrial carry domain; (B) the diagram of human MTCH2, yellow refers to the single middle mitochondrial carry domain from positions 123 to 206 (coloured in yellow); (C) 3D structure of human MTCH2. Note. (A and B) are adapted from “Mitochondrial carrier homolog 2 is a target of tBID in cells signaled to die by tumor necrosis factor alpha” by Michal Grinberg, Michal Schwarz, Yehudit Zaltsman, Tzipi Eini, Hagit Niv, Shmuel Pietrokovski & Atan Gross. Mol Cell Biol. 2005; 25: 4579–90.6 |
Structure Characteristics of MTCH2
MTCH2 was first identified as an uncharacterized protein of the putative open reading frames of CD34+ hematopoietic stem/progenitor cells by Zhu Chen8 and named because the single conserved mitochondrial carrier domain is similar to that contained in MTCH1. The MTCH family appears to have one mitochondrial carrier domain and three transmembrane domains with six transmembrane α-helices. Both the N and the C termini of MTCH2 are facing the inner membrane space (IMS), and the three long hydrophilic segments (connecting the two transmembrane regions of each domain) are facing the matrix (Figure 1).
The single conserved mitochondrial carrier domain in the MTCH family tends to have three trans-membrane domains. The unique mitochondrial carrier domain II allows the bind of the outside protein of mitochondria to the OMM but does not allow the transfer to the IMM, which is different from other mitochondrial carrier proteins binding outside proteins on the OMM and translocating them to the IMM.9 This is due to the hydrophilic segments of MTCH2 acting as a gatekeeper for the outer membrane, forming a prominent groove accessible to the charged and polar residues, whereas other proteins of the solute carrier 25 family which form a pore, allowing substrates to cross the membrane.1 MTCH2 is required for the biogenesis of diverse tail-anchored (TA) mitochondrial outer membrane proteins (ie, FUNDC1, CYB5B and VDAC1) and inserts α-helical proteins into mitochondria outer membrane, of which TA proteins are the largest class.1 A TA protein features a single C-terminal transmembrane domain. In contrast to other solute carrier 25 family members, MTCH2 has a sequence motif existing in both mitochondria and peroxisomes, and thus results in the mis-localisation of TA proteins to the endoplasmic reticulum.1 However, recent coarse-grained and atomistic simulations experiments revealed that MTCH2 substantially diminished or even destroyed the free energy barrier for lipid flip-flop, suggesting the high scrambling activity of MTCH2, similar to VDAC dimers.4
The error translation of MTCH2 has been found to occurr in the manner of a double stop codon read-through whose translation continues beyond the stop codon to a downstream stop codon based on the high translational readthrough efficiency of MTCH2. The double stop codon read-through of MTCH2 can be driven by a cis-acting RNA signal element, resulting in a highly unstable isoform with cytoplasmic localization, different from the canonical stable isoform and mitochondrial localisation.10
Apoptosis
The most well-known function of MTCH2 is in inducing apoptosis. MTCH2 binds with a pro-apoptotic B-cell lymphoma 2 family member cleaved BH3-interacting domain death agonist (cBID) to regulate the mitochondrial apoptosis. cBID consists of p7 and truncated BID (tBID). MTCH2 initially recruits cBID to mitochondria to unmask the BH3 domain of cBID, and then a complex of cBID, BAX and MTCH2 is built. The complex drives tBID to a highly extended conformation, and then the segments of 59–73 and 111–125 of tBID cross-link to 140–161 and 240–290 of MTCH2, respectively. Meanwhile, helices α 4–7 of tBID insert into the mitochondrial membrane.7,11–13 The deeper insertion of tBID is a prerequisite for formatting the complex of tBID and BAX, which forms lipidic pores allowing large macromolecules into mitochondria, leading to the increase in mitochondrial outer membrane permeabilization (MOMP) and release of multiple apoptogenic factors including cytochrome c. Cytochrome c released to cytosol sequentially activates effector caspases.6,7,14 Besides, the complex of the modulator of apoptosis-1 (MOAP-1) and MTCH2 has also been shown to be required to insert tBID into mitochondrion.15
The knockout of MTCH2 delays instead of eliminates the tBID triggered MOMP and apoptosis accompanied with the decreased sensitivity of the insertion of tBID to mitochondria.5,7,16 This could be because of the compensatory role of cardiolipin, which has a similar function to MTCH2 in activating tBID.17 An impaired recruitment of tBID was found in the depletion of both cardiolipin and MTCH2, but not in knockout cardiolipin or MTCH2 alone.16 Concordantly, overexpressed MTCH2 is markedly sensitive to apoptosis induced by imatinib treatment in human myelogenous leukemia K562 cells.1,16
Metabolic Diseases
On the basis of the two-fold increased mRNA of MTCH2 in human skeletal muscle in response to insulin that was first found in 200318 and the rapid development of single nucleotide polymorphisms (SNPs) detection technology, studies on the relevance of MTCH2 SNPs to obesity and diabetes emerged. Also rs10838738 is a cis-expressed quantitative trait locus on the expression of MTCH2 that has gained great attention. Dozens of studies revealed the association of rs10838738 SNP in MTCH2 with obesity and diabetes on the basis of the highly expressed MTCH2 in human white adipose tissue and adipocytes.19–22 However, the results of association between SNPs in or near MTCH2 with obesity and diabetes vary on population and age.23 Ethnicity has been found to be the most significant moderator affecting the heterogeneity for MTCH2 with BMI.24 An interaction between sex and MTCH2 SNPs among Chinese obesities was discovered.25 Moreover, the DNA methylation level of MTCH2 in human adipose tissue was found to be correlated with increased BMI,26 and played a prominent role with MTCH2 in adipogenesis in intramuscular fat regulation via regulation of the level of m6A-YTHDF1.27
The investigation in SNPs of MTCH2 is sequentially extended to other metabolic diseases. The significant associations with lower high density cholesterol levels,28 high blood pressure,29 metabolic syndrome,30 and polycystic ovary syndrome31 were observed, and these could be due to the enriched SNPs in or near MTCH2 in purine, deoxynucleotide, and ribo-deoxynucleotide metabolism32 and associated with total carbohydrate, mono- and disaccharide, and polysaccharide intakes.33 There is a non-referent allele, MTCH2p.Pro290Ala, which is also found to be associated with increased BMI, and the referent allele of p.Prp 290 (rs1064608) in MTCH2 induces the lower expression of MTCH2.34
The relation between SNPs of MTCH2 and metabolism has been found in child growth and development, i.e., lower birth weight35 and childhood BMI trajectories.36 However, the association with paediatric BMI is controversial,37,38 and no association with BMI across adulthood is observed.39 The genetic risk score built by summing risk alleles including MTCH2 SNPs is found to be related to BMI, obesity, faster weight gain in childhood40 and early menarche.41
The relation between MTCH2 and metabolism could be attributable to the involvement of MTCH2 in lipid homeostasis/storage and glucose metabolism. Homozygous mutants of Mtch1 in C. elegans. significantly reduce fat storage, and overexpression of Mtch1 triggers lipid accumulation.42 The knockdown of Mtch2 in Drosophila results in cardiomyopathy, less adiposity and a shortened life span.43 Consistently, Mtch2-GFP transgenic mice and MTCH2 overexpressed HEK29 cells show the dysfunction of lipid metabolism, resulting in fatty and lactate accumulation and impaired oxygen consumption, and this might be regulated by the increased pyruvate dehydrogenase kinase, genes of medium chain acyl-CoA dehydrogenase, acetyl-CoA acyltransferase 2 and fatty acid synthase (FASN).43,44 However, MTCH2 is favourable in the situation where glucose oxidation and tricarboxylic acid cycle are predominant, such as heart failure.43 Moreover, MTCH2 is helpful in stimulating mitochondrial hyperfusion to elongate mitochondria in a bioactive lipogenesis intermediated lysophosphatidic acid way to boost energy production under starving conditions.45 The direct binding between estrogen receptor alpha (ERa) and MTCH2 has been identified by a yeast-two-hybrid screen,46 and a further finding revealed that MTCH2 has an inhibitive impact on the transcriptional potential of ERa, a known regulator of metabolic homeostasis.42
The effect of MTCH2 targeted clinical assessment and potential intervention have been assessed. The genetic risk scores composed of seven SNPs including MTCH2 that is negatively associated with excess BMI loss after roux-en-Y gastric bypass surgery.47 However, weight loss among obese women does not alter the expression of MTCH2,19 and alleles of MTCH2 are not associated with weight regain in the course of one year after a lifestyle intervention in overweight children.48 Mtch2 deletion mice eat more compared to control mice when fed a high-fat diet but are less susceptible to weight gain with less fat per body weight and lower levels of circulating insulin.49 This could be due to the raised muscle metabolic capacity, energy demand and expenditure resulting from the increased mitochondrial metabolism and mass.49 Superparamagnetic iron oxide nanoparticles induce the upregulation of MTCH2 in human adipocytes among diabetics,50 which might be helpful for the apoptosis of adipocytes. The MTCH2–treatment interactions are observed for short-term weight loss among diabetics.51 Moreover, a lower expression of MTCH2 has been found in eclampsia patients, but the underlying mechanism was not investigated.52
Neurodegenerative Diseases
The highest MTCH2 expression level is in the hippocampal formation and cerebellum in the mouse brain.53 Decreased gene expression of MTCH2 has been identified as a risk factor of Alzheimer’s disease (AD).54,55 SNPs in MTCH2 have been shown to be related to AD,32,56 schizophrenia57 and emotional eating in humans.58 Functional variants at 11p11.2 of MTCH2 is involved in AD development via altering cellular amyloid-b and phosphorylated tau.59 MTCH2 is a conjointed gene, in which SNPs are related to both cardiovascular and AD,60 both obesity and AD,56 and both AD and glaucoma.61
The underlying mechanism of the relation between MTCH2 and AD seems to be related to the mitochondrial apoptosis and the interaction with other AD genes. Mitochondria apoptosis that regulated by cooperation between MTCH2 with BID promotes Ca2+-induced neuronal injury.62 Mtch2 knockout in the mouse forebrain triggers the deficiency of mitochondria calcium handling and mitochondrial motility in the hippocampus, resulting in the reduction of synaptic connectivity, motor coordination and impaired hippocampal long-term potentiation and rotarod test.63 Meanwhile, activated microglia was increased, and neuron density was reduced.64 However, the specific influence of MTCH2 on neurodegenerative disorders varies with age.64 Mtch2 deficient 3-month-old mice have an increased swimming speed compared with controls, but Mtch2 forebrain conditional knockout mice at the ages of 9 and 14 months have a deterioration of spatial navigation and lower short- and long-term potentiation accompanied with plastic properties and neurodegeneration in the hippocampus.64 There is a shorter stay in the rotarod test in MTCH2 conditional knockout mice at age 3 months than at ages 9 and 14 months. The electrophysiological response to the tetanic stimulation is significantly different between Mtch2 forebrain conditional knockout mice at the ages of 9 and 14 months. There is a contrast result in Caenorhabditis elegans, in which age-related proteostasis collapse is suppressed by the loss of MTCH1/MTCH2 via activating CED-4/3 and heat shock proteins and suppressing the activity of HSF-1.65
MTCH2 has been found to be associated with AD related genes or mental diseases related genes, including CELF1,55 SPI132 and FMR1.66 CELF1 is an AD risk gene, and there is expression of MTCH2 within CELF1 locus in AD brains.55 MTCH2 is associated with SPI1, a well-known microglial transcription factor. SPI1 also interacts with CELF1, and the knockdown of SPI1 reduces AD pathology.32 FMR1 encodes fragile X mental retardation proteins, loss of which is the leading cause of inherited forms of mental retardation and autism, and MTCH2 is negatively correlated to the level of FMR1.66 SNPs of MTCH2 are related to emotional eating in humans64 but not in zebrafish.67
Cancers
MTCH2 has been identified as a “Poised Gene Cassette” to balance the complex interplay of pro- and anti-oncogenic genes,68 and the role of MTCH2 is different in different cancers. Inhibition of MTCH2 tends to have therapeutic effects on acute myeloid leukemia (AML), glioblastomas, hepatocellular carcinoma and osteosarcoma, but seems to deteriorate colorectal cancer, cervical cancer and gastric cancer. In AML, inhibition of MTCH2 shows the loss of engraftment potential effect in vitro and in vivo.69 The knockout of MTCH2 depresses growth, viability and clonogenic growth in AML cells.69 Moreover, MTCH2 knockdown promotes differentiation from hematopoietic stem cells to differentiated myeloid population and from primitive/stem-like AML fractions to differentiated AML cells.69 Mtch2 knockout in a murine model of leukemia transduced with Mll-af9 oncogene decreases the leukemogenic potential of MLL-AF9 oncogene,69 and this might be because of the increased histone acetylation that resulted from the increased mitochondrial/nuclear pyruvate content and pyruvate dehydrogenase instead of the pro-apoptotic effect of BID combination.69
A MTCH2 (Chr11: 47,647,265 A>G) gene sequence change is found in rapidly growing glioblastoma, indicating that the change of variant prioritization after annotation may be involved in the aggression of glioblastomas.70 The high expression of MTCH2 in malignant glioma has been shown to be related to a metabolic shift from OXPHO to aerobic glycolysis and mitochondrial dysfunctions, and AKT signaling seems to be involved in this relationship.71 Mitochondrial dysfunction and apoptosis in hepatocellular carcinoma have been shown to be related to aberrantly enriched nuclear-encoded long noncoding RNA (lncRNA) metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), and the transportation of MALAT1 to mitochondria is mediated by MTCH2.72 Upregulated MTCH2 has also been found in clinical osteosarcoma samples.73 In osteosarcoma, overexpression of MTCH2 rescues the attenuated cell viability, inhibited proliferation, promoted cell apoptosis, elevated OXPHOS and suppressed the Warburg effect triggered by APOC1 silencing,74 and the regulation of oncogene FASN on MTCH2 is detected by iTRAQ-based proteomic analysis.75
On the contrary, upregulation of MTCH2 tends to have a therapeutic effect on thymic squamous cell carcinomas, cervical cancer, colorectal cancer and gastric cancer. The therapeutic effect of MTCH2 in thymic squamous cell carcinomas is through inducing apoptosis.76 MTCH2 has been found to be a therapeutic target of dihydroartemisin and alisertib. Upregulated MTCH2 and caveolin 1, after dihydroartemisin treatment, inhibit cell viability and prompt the apoptosis of cervical cancer cells and increases the sensitivity to treatment.77 A network pharmacology analysis reveals that MTCH2 is a potential therapeutic target of alisertib to treat colorectal cancer,78 and SNPs of MTCH2 have been found to be related with colorectal cancer.79 Further depletion of both cardiolipin and MTCH2 results in an impaired recruitment of tBID in the human colorectal cancer cell line HCT116.16 Similarly, silence of MTCH2 increases the invasion of gastric cancer cells.68 MTCH2 cooperates with the mitochondrial ubiquitin E3 ligase MARCH5 and the ubiquitin E2 conjugating enzyme UBE2K to degrade myeloid cell leukemia-1 protein (MCL1) by the proteasome.80 This facilitates the clearance of unfit cells following DNA damage and cell cycle arrest in many types of cancer cells.80 Moreover, treatment with aminoethyl dihydrogen phosphate upregulates the protein expression of MTCH2, BAD and BAX and downregulates antiapoptotic protein B-cell lymphoma 2.81 Bioinformatics analysis reveals the association between MTCH2 and tumor-promoting factors and the alteration of the DNA methylation pattern of MTCH2 in different cancer tissues, moreover, the combined effect of clustered genes, including MTCH2, could predict the survival probability of patients with 14 types of cancer.82
The findings of the effect of MTCH2 on breast cancer are controversial, and this might be due to the fact that breast cancer has diverse sub-types and different stages. Most coding sequences of MTCH2 are conjoint with exons 7 to 17 of FNBP4 to construct a fusion gene, which has been found to be associated with breast cancer83 and myofibroma.84 The decreased mRNA expression of MTCH2 is reverse regulated by abundant miR-135b in basal and normal-like human breast cancers.85 Similarly, the involvement of MTCH2 in normal human breast gland development might result in the association of MTCH2 with the epithelial–mesenchymal transition in breast cancer.86 However, the protein level of MTCH2 is increased 2.3 times in breast tumor MDA-MB-231 cell line after treatment with sub-toxic dose of snake venom compared with untreated cells.87 MTCH2 expression reduces Met-HGF/SF-induced proliferation and scattering by attenuating and altering the downstream signaling of Met in breast cancer cell lines.73 SNPs of MTCH2 have also been found to be related with endometrial cancer.88
Embryonic Development
MTCH2 is necessary for embryogenesis and plays a crucial role in fertility, formation of liver and intestine and differentiation of haematopoietic stem cells. Although heterozygote Mtch2± mice are viable, fertile and show no phenotypic abnormalities, homozygous Mtch2−/− mice die at embryonic day 7.5 due to multiple defects, suggesting that MTCH2 is a prerequisite for embryogenesis.5 Knockdown of Mtch2 progressively affects postembryonic growth of zebrafish larvae with significantly smaller embryo than control zebrafish at 8dpf and the impaired development of liver and intestine, and this could be resulted from metabolic impairment and an energy deficient state.67
MTCH2 helps to maintain the self-renewal and quiescence of haematopoietic stem cells (HSCs) in bone marrow niches89 and in embryonic stem cells (ESCs).90 Non-motile HSCs enter into peripheral cycle replenishment with the blood with mature leukocytes and red blood cells on demand, leading to exhaustion of quiescent HSCs. Loss of MTCH2 facilitates the transition of non-motile HSC into cycles via enhancing the proliferation of HSCs, and this relates to the metabolic shift from glycolysis to OXPHOS in HSCs, where glycolysis is preferred and the enlarged mitochondria.89,91 Loss of MTCH2 and elevation in OXPHOS have been shown to protect HSCs and haematopoietic progenitor cells from irradiation-induced death in vivo and in intro.89 MTCH2 prompts mitochondrial fusion and depresses mitochondrial fission in the naïve-to-primed interconversion during embryo development, resulting in mitochondria elongation and an increase in mitochondrial mass, mtRNA and mitochondria respiration.91,92 The elongated mitochondria facilitate partial exit from naive pluripotency without entering differentiation via modulating nuclear gene reprogramming to drive reprogramming of mitochondria metabolism including glucose and glutamine metabolism and histone deacetylation.91 In ESCs, overexpression MTCH2 rescues the COPS5 knockout induced growth defect, reduced glycolysis, enhanced OXPHOS, elevated reactive oxygen species (ROS) and subsequent DNA damage accumulation,90 suggesting that MTCH2 facilitates ESC maintaining genome integrity and self-renewal capacity. The translation of the MTCH2 gene or MTCH2-regulated mitochondria from bone marrow mesenchymal stem cells to RAW264.7 cells, which suppresses the upregulated apoptosis and ROS stimulated by lipopolysaccharide,93 suggesting the potential for MTCH2 treatment.
Reproduction
The testis is an organ in a high level of apoptosis with the expression of MTCH2, which is initially detected during the first spermatogenic wave and fluctuates depending on the different developmental stages and testicular cells.94 MTCH2 level increases with mild hyperthermia-induced testicular apoptosis.94 Similarly, the Mtch2 overexpression mouse line shows a low fertility rate and no progeny for the last generation.44 On the contrary, the reduction of Mtch1 in worms results in fertility defects, homozygous mutants of Mtch1 in C. elegans. and MTCH2 in mice cause sterile adults,42 and loss of MTCH2 is detected in cattle-yak.95 These findings suggest that an appropriate level of MTCH2 is required for male fertility. A recent study reveals that the level of MTCH2 expression and subsequent pro-apoptotic proteins including BID, BAX, cytochrome C, caspase-9, and caspase-3 are decreased after the stimulation of silica nanoparticles, resulting in the apoptosis of spermatogenic cells.96
Discussion
The first well-known role of MTCH2 was regulating apoptosis, and this affected the following studies. The vast quantities of SNP studies investigate the associations of MTCH2 with metabolic diseases including obesity, diabetes, cardiovascular diseases and neurodegenerative diseases including AD, leading to the further exploration of the underlying mechanism of the association. Due to the high metabolic demand and suppressed apoptosis of cancer cells, upregulation of MTCH2 tends to have a therapeutic effect on colorectal cancer, cervical cancer and gastric cancer. However, inhibition of MTCH2 shows the therapeutic prospect on several cancers, i.e., the myeloid leukemia, glioblastomas and hepatocellular carcinoma. With the application of technology of gene knockout in mouse, the fatality of homozygous Mtch2−/− mice attracts attentions on the role of MTCH2 in embryonic development. MTCH2 is found to be a prerequisite of embryogenesis and organ development (i.e., liver and intestine), a protector of self-renewal and quiescence of stem cells and a promotor of spermatogenesis.
Studies on the function of MTCH2 in health and diseases have been increasingly emerging, however, the current findings generally are still in the early stages. There are many research fields that MTCH2 has not been considered for, such as follicular development, pregnancy complications, immunological diseases and respiration system diseases, etc. A sign of the therapeutic potential of MTCH2 has emerged based on the fact that superparamagnetic iron oxide nanoparticles, dihydroartemisin, alisertib, aminoethyl dihydrogen phosphate and snake venom could regulate the expression of MTCH2, and moreover MTCH2 seems to be the downstream factor of FASN, COPS5 and miR-150. However, none of the above studies is initially designed to investigate that MTCH2 acts as a treatment target through excluding the interference with other molecules.
In conclusion, the emerging studies demonstrate the physiological role of MTCH2 in metabolic diseases, neurodegenerative diseases, cancers, embryonic development and reproduction, although further investigations for deeper elucidation are required. This review fills the gap in the summary of the current findings with MTCH2, from disease-oriented aspects, to highlighting the therapeutic prospect of MTCH2 and encouraging future studies to discover more comprehensive functions of MTCH2 in health and diseases.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
XP received a National Natural Science Foundation of China (NSFC-82000399) and YC received a research grant from the National Key Research and Development Program of China (2022YFC2703000).
Disclosure
All the authors declare that they do not have any financial and personal relationships with other people or organizations that could influence the design, supervision or reporting of their work. The authors declare that they have no competing interests.
References
1. Guna A, Stevens TA, Inglis AJ, et al. MTCH2 is a mitochondrial outer membrane protein insertase. Science. 2022;378(6617):317–322. doi:10.1126/science.add1856
2. Li D, Schilling -R-RC, Reinisch MA, Vanni KM. Lipid scrambling is a general feature of protein insertases. bioRxiv. doi:10.1101/2023.09.01.555937
3. Alexander Brandis TM, Malitsky S, Itkin M, et al. High-energy demand and nutrient exhaustion in MTCH2 knockout cells. bioRxiv Preprint. 2023.
4. Bartoš L, Vácha MA, Vácha R. Insertases Scramble Lipids: molecular Simulations of MTCH2. bioRxiv. 2023. doi:10.1101/2023.08.14.553169
5. Zaltsman Y, Shachnai L, Yivgi-Ohana N, et al. MTCH2/MIMP is a major facilitator of tBID recruitment to mitochondria. Nat Cell Biol. 2010;12(6):553–562. doi:10.1038/ncb2057
6. Grinberg M, Schwarz M, Zaltsman Y, et al. Mitochondrial carrier homolog 2 is a target of tBID in cells signaled to die by tumor necrosis factor alpha. Mol Cell Biol. 2005;25(11):4579–4590. doi:10.1128/MCB.25.11.4579-4590.2005
7. Shamas-Din A, Bindner S, Zhu W, et al. tBid undergoes multiple conformational changes at the membrane required for Bax activation. J Biol Chem. 2013;288(30):22111–22127. doi:10.1074/jbc.M113.482109
8. Zhang QH, Ye M, Wu XY, et al. Cloning and functional analysis of cDNAs with open reading frames for 300 previously undefined genes expressed in CD34+ hematopoietic stem/progenitor cells. Genome Res. 2000;10(10):1546–1560. doi:10.1101/gr.140200
9. Schwarz M, Andrade-Navarro MA, Gross A. Mitochondrial carriers and pores: key regulators of the mitochondrial apoptotic program? Apoptosis. 2007;12(5):869–876. doi:10.1007/s10495-007-0748-2
10. Manjunath LE, Singh A, Sahoo S, et al. Stop codon read-through of mammalian MTCH2 leading to an unstable isoform regulates mitochondrial membrane potential. J Biol Chem. 2020;295(50):17009–17026. doi:10.1074/jbc.RA120.014253
11. Veresov VG, Davidovskii AI. Structural insights into proapoptotic signaling mediated by MTCH2, VDAC2, TOM40 and TOM22. Cell Signal. 2014;26(2):370–382. doi:10.1016/j.cellsig.2013.11.016
12. Hung CL, Chang HH, Lee SW, et al. Stepwise activation of the pro-apoptotic protein Bid at mitochondrial membranes. Cell Death Differ. 2021;28(6):1910–1925. doi:10.1038/s41418-020-00716-5
13. Katz C, Zaltsman-Amir Y, Mostizky Y, et al. Molecular basis of the interaction between proapoptotic truncated Bid (tBID) protein and mitochondrial carrier homologue 2 (MTCH2) protein: key players in mitochondrial death pathway. J Biol Chem. 2012;287(18):15016–15023. doi:10.1074/jbc.M111.328377
14. Shamas-Din A, Satsoura D, Khan O, et al. Multiple partners can kiss-and-run: bax transfers between multiple membranes and permeabilizes those primed by tBid. Cell Death Dis. 2014;5(6):e1277. doi:10.1038/cddis.2014.234
15. Tan CT, Zhou QL, Su YC, et al. MOAP-1 mediates fas-induced apoptosis in liver by facilitating tbid recruitment to mitochondria. Cell Rep. 2016;16(1):174–185. doi:10.1016/j.celrep.2016.05.068
16. Raemy E, S, Montessuit S, Pierredon S, van Kampen AH, Vaz FM, Martinou J-C. Cardiolipin or MTCH2 can serve as tBID receptors during apoptosis. Cell Death Differ. 2016;23(7):1165–1174. doi:10.1038/cdd.2015.166
17. Shamas-Din A, Bindner S, Chi X, et al. Distinct lipid effects on tBid and Bim activation of membrane permeabilization by pro-apoptotic Bax. Biochem J. 2015;467(3):495–505. doi:10.1042/BJ20141291
18. Rome S, Clement K, Rabasa-Lhoret R, et al. Microarray profiling of human skeletal muscle reveals that insulin regulates approximately 800 genes during a hyperinsulinemic clamp. J Biol Chem. 2003;278(20):18063–18068. doi:10.1074/jbc.M300293200
19. Kulyté A, Rydén M, Mejhert N, et al. MTCH2 in human white adipose tissue and obesity. J Clin Endocrinol Metab. 2011;96(10):E1661–5. doi:10.1210/jc.2010-3050
20. Bernhard F, Landgraf K, Klöting N, et al. Functional relevance of genes implicated by obesity genome-wide association study signals for human adipocyte biology. Diabetologia. 2013;56(2):311–322. doi:10.1007/s00125-012-2773-0
21. Ng MC, Tam CH, So WY, et al. Implication of genetic variants near NEGR1, SEC16B, TMEM18, ETV5/DGKG, GNPDA2, LIN7C/BDNF, MTCH2, BCDIN3D/FAIM2, SH2B1, FTO, MC4R, and KCTD15 with obesity and type 2 diabetes in 7705 Chinese. J Clin Endocrinol Metab. 2010;95(5):2418–2425. doi:10.1210/jc.2009-2077
22. He M, Cornelis MC, Franks PW, et al. Obesity genotype score and cardiovascular risk in women with type 2 diabetes mellitus. Arterioscler Thromb Vasc Biol. 2010;30(2):327–332. doi:10.1161/ATVBAHA.109.196196
23. Graff M, North KE, Richardson AS, et al. BMI loci and longitudinal BMI from adolescence to young adulthood in an ethnically diverse cohort. Int J Obes Lond. 2017;41(5):759–768. doi:10.1038/ijo.2016.233
24. Pei YF, Tian Q, Zhang L, et al. Exploring the major sources and extent of heterogeneity in a genome-wide association meta-analysis. Ann Hum Genet. 2016;80(2):113–122. doi:10.1111/ahg.12143
25. Kong X, Xing X, Zhang X, et al. Sexual Dimorphism of a Genetic Risk Score for Obesity and Related Traits among Chinese Patients with Type 2 Diabetes. Obes Facts. 2019;12(3):328–343. doi:10.1159/000500490
26. Rönn T, Volkov P, Gillberg L, et al. Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum Mol Genet. 2015;24(13):3792–3813. doi:10.1093/hmg/ddv124
27. Jiang Q, Sun B, Liu Q, et al. MTCH2 promotes adipogenesis in intramuscular preadipocytes via an m(6)A-YTHDF1-dependent mechanism. FASEB j. 2019;33(2):2971–2981. doi:10.1096/fj.201801393RRR
28. van Vliet-Ostaptchouk JV, den Hoed M, Luan J, et al. Pleiotropic effects of obesity-susceptibility loci on metabolic traits: a meta-analysis of up to 37,874 individuals. Diabetologia. 2013;56(10):2134–2146. doi:10.1007/s00125-013-2985-y
29. Melka MG, Bernard M, Mahboubi A, et al. Genome-wide scan for loci of adolescent obesity and their relationship with blood pressure. J Clin Endocrinol Metab. 2012;97(1):E145–50. doi:10.1210/jc.2011-1801
30. Hotta K, Kitamoto T, Kitamoto A, et al. Association of variations in the FTO, SCG3 and MTMR9 genes with metabolic syndrome in a Japanese population. J Hum Genet. 2011;56(9):647–651. doi:10.1038/jhg.2011.74
31. Louwers YV, Rayner NW, Herrera BM, et al. BMI-associated alleles do not constitute risk alleles for polycystic ovary syndrome independently of BMI: a case-control study. PLoS One. 2014;9(1):e87335. doi:10.1371/journal.pone.0087335
32. Gockley J, Montgomery KS, Poehlman WL, et al. Multi-tissue neocortical transcriptome-wide association study implicates 8 genes across 6 genomic loci in Alzheimer’s disease. Genome Med. 2021;13(1):76. doi:10.1186/s13073-021-00890-2
33. Bauer F, Elbers CC, Adan RA, et al. Obesity genes identified in genome-wide association studies are associated with adiposity measures and potentially with nutrient-specific food preference. Am J Clin Nutr. 2009;90(4):951–959. doi:10.3945/ajcn.2009.27781
34. Turcot V, Lu Y, Highland HM, et al. Protein-altering variants associated with body mass index implicate pathways that control energy intake and expenditure in obesity. Nat Genet. 2018;50(1):26–41. doi:10.1038/s41588-017-0011-x
35. Kilpeläinen TO, den Hoed M, Ong KK, et al. Obesity-susceptibility loci have a limited influence on birth weight: a meta-analysis of up to 28,219 individuals. Am J Clin Nutr. 2011;93(4):851–860. doi:10.3945/ajcn.110.000828
36. Warrington NM, Wu YY, Pennell CE, et al. Modelling BMI trajectories in children for genetic association studies. PLoS One. 2013;8(1):e53897. doi:10.1371/journal.pone.0053897
37. Zhao J, Bradfield JP, Li M, et al. The role of obesity-associated loci identified in genome-wide association studies in the determination of pediatric BMI. Obesity. 2009;17(12):2254–2257. doi:10.1038/oby.2009.159
38. Wang J, Mei H, Chen W, et al. Study of eight GWAS-identified common variants for association with obesity-related indices in Chinese children at puberty. Int J Obes Lond. 2012;36(4):542–547. doi:10.1038/ijo.2011.218
39. Graff M, Gordon-Larsen P, Lim U, et al. The influence of obesity-related single nucleotide polymorphisms on BMI across the life course: the PAGE study. Diabetes. 2013;62(5):1763–1767. doi:10.2337/db12-0863
40. Elks CE, Loos RJ, Hardy R, et al. Adult obesity susceptibility variants are associated with greater childhood weight gain and a faster tempo of growth: the 1946 British Birth Cohort Study. Am J Clin Nutr. 2012;95(5):1150–1156. doi:10.3945/ajcn.111.027870
41. Mumby HS, Elks CE, Li S, et al. Mendelian randomisation study of childhood BMI and early menarche. J Obes. 2011;2011:180729. doi:10.1155/2011/180729
42. Rottiers V, Francisco A, Platov M, et al. MTCH2 is a conserved regulator of lipid homeostasis. Obesity. 2017;25(3):616–625. doi:10.1002/oby.21751
43. Fischer JA, Monroe TO, Pesce LL, et al. Opposing effects of genetic variation in MTCH2 for obesity versus heart failure. Hum Mol Genet. 2023;32(1):15–29. doi:10.1093/hmg/ddac176
44. Bar-Lev Y, Moshitch-Moshkovitz S, Tsarfaty G, et al. Mimp/Mtch2, an Obesity Susceptibility Gene, Induces Alteration of Fatty Acid Metabolism in Transgenic Mice. PLoS One. 2016;11(6):e0157850. doi:10.1371/journal.pone.0157850
45. Labbé K, Mookerjee S, Le Vasseur M, et al. The modified mitochondrial outer membrane carrier MTCH2 links mitochondrial fusion to lipogenesis. J Cell Biol. 2021;220(11). doi:10.1083/jcb.202103122
46. Albers M, Kranz H, Kober I, et al. Automated yeast two-hybrid screening for nuclear receptor-interacting proteins. Mol Cell Proteomics. 2005;4(2):205–213. doi:10.1074/mcp.M400169-MCP200
47. Bandstein M, Voisin S, Nilsson EK, et al. A genetic risk score is associated with weight loss following roux-en y gastric bypass surgery. Obes Surg. 2016;26(9):2183–2189. doi:10.1007/s11695-016-2072-9
48. Hinney A, Wolters B, Pütter C, et al. No impact of obesity susceptibility loci on weight regain after a lifestyle intervention in overweight children. J Pediatr Endocrinol Metab. 2013;26(11–12):1209–1213. doi:10.1515/jpem-2013-0179
49. Buzaglo-Azriel L, Kuperman Y, Tsoory M, et al. Loss of muscle mtch2 increases whole-body energy utilization and protects from diet-induced obesity. Cell Rep. 2016;14(7):1602–1610. doi:10.1016/j.celrep.2016.01.046
50. Sharifi S, Daghighi S, Motazacker MM, et al. Superparamagnetic iron oxide nanoparticles alter expression of obesity and T2D-associated risk genes in human adipocytes. Sci Rep. 2013;3:2173. doi:10.1038/srep02173
51. Delahanty LM, Pan Q, Jablonski KA, et al. Genetic predictors of weight loss and weight regain after intensive lifestyle modification, metformin treatment, or standard care in the Diabetes Prevention Program. Diabetes Care. 2012;35(2):363–366. doi:10.2337/dc11-1328
52. Shi Z, Long W, Zhao C, et al. Comparative proteomics analysis suggests that placental mitochondria are involved in the development of pre-eclampsia. PLoS One. 2013;8(5):e64351. doi:10.1371/journal.pone.0064351
53. Lein ES, Hawrylycz MJ, Ao N, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445(7124):168–176. doi:10.1038/nature05453
54. Escott-Price V, Bellenguez C, Wang LS, et al. Gene-wide analysis detects two new susceptibility genes for Alzheimer’s disease. PLoS One. 2014;9(6):e94661. doi:10.1371/journal.pone.0094661
55. Karch CM, Ezerskiy LA, Bertelsen S, et al. Alzheimer’s Disease Risk Polymorphisms Regulate Gene Expression in the ZCWPW1 and the CELF1 Loci. PLoS One. 2016;11(2):e0148717. doi:10.1371/journal.pone.0148717
56. Zhuang QS, Zheng H, Gu XD, et al. Detecting the genetic link between Alzheimer’s disease and obesity using bioinformatics analysis of GWAS data. Oncotarget. 2017;8(34):55915–55919. doi:10.18632/oncotarget.19115
57. Purcell SM, Moran JL, Fromer M, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506(7487):185–190. doi:10.1038/nature12975
58. Cornelis MC, Rimm EB, Curhan GC, et al. Obesity susceptibility loci and uncontrolled eating, emotional eating and cognitive restraint behaviors in men and women. Obesity (Silver Spring. 2014;22(5):E135–41. doi:10.1002/oby.20592
59. Xu M, Liu Q, Bi R, et al. Co-existence of multiple functional variants and genes underlie genetic risk locus 11p11.2 of Alzheimer’s disease. Biol Psychiatry. 2023;94:743–759. doi:10.1016/j.biopsych.2023.05.020
60. Broce IJ, Tan CH, Fan CC, et al. Dissecting the genetic relationship between cardiovascular risk factors and Alzheimer’s disease. Acta Neuropathol. 2019;137(2):209–226. doi:10.1007/s00401-018-1928-6
61. Zheng C, Liu S, Zhang X, et al. Shared genetic architecture between the two neurodegenerative diseases: alzheimer’s disease and glaucoma. Front Aging Neurosci. 2022;14:880576. doi:10.3389/fnagi.2022.880576
62. D’Orsi B, Niewidok N, Düssmann H, et al. Mitochondrial carrier homolog 2 functionally co-operates with bh3 interacting-domain death agonist in promoting Ca(2+)-Induced Neuronal Injury. Front Cell Dev Biol. 2021;9:750100. doi:10.3389/fcell.2021.750100
63. Ruggiero A, Aloni E, Korkotian E, et al. Loss of forebrain MTCH2 decreases mitochondria motility and calcium handling and impairs hippocampal-dependent cognitive functions. Sci Rep. 2017;7:44401. doi:10.1038/srep44401
64. Aloni E, Ruggiero A, Gross A, et al. Learning deficits in adult mitochondria carrier homolog 2 forebrain knockout mouse. Neuroscience. 2018;394:156–163. doi:10.1016/j.neuroscience.2018.10.035
65. Aman Y, Erinjeri AP, Tataridas-Pallas N, et al. Loss of MTCH-1 suppresses age-related proteostasis collapse through the inhibition of programmed cell death factors. Cell Rep. 2022;41(8):111690. doi:10.1016/j.celrep.2022.111690
66. Xu B, Zhang Y, Zhan S, et al. proteomic profiling of brain and testis reveals the diverse changes in ribosomal proteins in fmr1 knockout mice. Neuroscience. 2018;371:469–483. doi:10.1016/j.neuroscience.2017.12.023
67. Landgraf K, Strobach A, Kiess W, et al. Loss of mtch2 function impairs early development of liver, intestine and visceral adipocytes in zebrafish larvae. FEBS Lett. 2016;590(17):2852–2861. doi:10.1002/1873-3468.12330
68. Yu K, Ganesan K, Tan LK, et al. A precisely regulated gene expression cassette potently modulates metastasis and survival in multiple solid cancers. PLoS Genet. 2008;4(7):e1000129. doi:10.1371/journal.pgen.1000129
69. Khan DH, Mullokandov M, Wu Y, et al. Mitochondrial carrier homolog 2 is necessary for AML survival. Blood. 2020;136(1):81–92. doi:10.1182/blood.2019000106
70. Khan I, Işık EB, Mahfooz S, et al. Identification of genetic alterations in rapid progressive glioblastoma by use of whole exome sequencing. Diagnostics. 2023;13(6).
71. Yuan Q, Yang W, Zhang S, et al. Inhibition of mitochondrial carrier homolog 2 (MTCH2) suppresses tumor invasion and enhances sensitivity to temozolomide in malignant glioma. Mol Med. 2021;27(1):7. doi:10.1186/s10020-020-00261-4
72. Zhao Y, Zhou L, Li H, et al. Nuclear-Encoded lncRNA MALAT1 Epigenetically Controls Metabolic Reprogramming in HCC Cells through the Mitophagy Pathway. Mol Ther Nucleic Acids. 2021;23:264–276. doi:10.1016/j.omtn.2020.09.040
73. Robinson AJ, Kunji ER, Gross A. Mitochondrial carrier homolog 2 (MTCH2): the recruitment and evolution of a mitochondrial carrier protein to a critical player in apoptosis. Exp Cell Res. 2012;318(11):1316–1323. doi:10.1016/j.yexcr.2012.01.026
74. Li R, He H, He X. APOC1 promotes the progression of osteosarcoma by binding to MTCH2. Exp Ther Med. 2023;25(4):163. doi:10.3892/etm.2023.11862
75. Fu D, Liu S, Liu J, et al. iTRAQ-based proteomic analysis of the molecular mechanisms and downstream effects of fatty acid synthase in osteosarcoma cells. J Clin Lab Anal. 2021;35(3):e23653. doi:10.1002/jcla.23653
76. Huang B, Belharazem D, Li L, et al. Anti-apoptotic signature in thymic squamous cell carcinomas - functional relevance of anti-apoptotic birc3 expression in the thymic carcinoma cell line 1889c. Front Oncol. 2013;3:316. doi:10.3389/fonc.2013.00316
77. Zhang T, Hu Y, Wang T, et al. Dihydroartemisinin inhibits the viability of cervical cancer cells by upregulating caveolin 1 and mitochondrial carrier homolog 2: involvement of p53 activation and NAD(P)H:quinone oxidoreductase 1 downregulation. Int J Mol Med. 2017;40(1):21–30. doi:10.3892/ijmm.2017.2980
78. Ren BJ, Zhou ZW, Zhu DJ, et al. Alisertib Induces Cell Cycle Arrest, Apoptosis, Autophagy and Suppresses EMT in HT29 and Caco-2 Cells. Int J Mol Sci. 2015;17(1):41. doi:10.3390/ijms17010041
79. Lim U, Wilkens LR, Monroe KR, et al. Susceptibility variants for obesity and colorectal cancer risk: the multiethnic cohort and PAGE studies. Int J Cancer. 2012;131(6):E1038–43. doi:10.1002/ijc.27592
80. Djajawi TM, Liu L, Gong JN, et al. MARCH5 requires MTCH2 to coordinate proteasomal turnover of the MCL1:NOXA complex. Cell Death Differ. 2020;27(8):2484–2499. doi:10.1038/s41418-020-0517-0
81. Conceição TO, Cabral L, Laveli-Silva MG, et al. New potential antiproliferative monophosphoester 2-aminoethyl dihydrogen phosphate in K-562 and K-562 MDR(+) leukemia cells. Biomed Pharmacother. 2021;142:112054. doi:10.1016/j.biopha.2021.112054
82. Huang T, Huang X, Nie Y, et al. A combined effect of expression levels of obesity-related genes and clinical factors on cancer survival rate. Biomed Res Int. 2020;2020:8838676. doi:10.1155/2020/8838676
83. Yoshihara K, Wang Q, Torres-Garcia W, et al. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene. 2015;34(37):4845–4854. doi:10.1038/onc.2014.406
84. Dachy G, Fraitag S, Boulouadnine B, et al. Novel COL4A1-VEGFD gene fusion in myofibroma. J Cell Mol Med. 2021;25(9):4387–4394. doi:10.1111/jcmm.16502
85. Arigoni M, Barutello G, Riccardo F, et al. miR-135b coordinates progression of ErbB2-driven mammary carcinomas through suppression of MID1 and MTCH2. Am J Pathol. 2013;182(6):2058–2070. doi:10.1016/j.ajpath.2013.02.046
86. Wang Q, Karvelsson ST, Johannsson F, et al. UDP-glucose dehydrogenase expression is upregulated following EMT and differentially affects intracellular glycerophosphocholine and acetylaspartate levels in breast mesenchymal cell lines. Mol Oncol. 2022;16(9):1816–1840. doi:10.1002/1878-0261.13172
87. Kisaki CY, Arcos SSS, Montoni F, et al. Bothrops Jararaca snake venom modulates key cancer-related proteins in breast tumor cell lines. Toxins. 2021;13(8):519. doi:10.3390/toxins13080519
88. Delahanty RJ, Beeghly-Fadiel A, Xiang YB, et al. Association of obesity-related genetic variants with endometrial cancer risk: a report from the Shanghai Endometrial Cancer Genetics Study. Am J Epidemiol. 2011;174(10):1115–1126. doi:10.1093/aje/kwr233
89. Maryanovich M, Zaltsman Y, Ruggiero A, et al. An MTCH2 pathway repressing mitochondria metabolism regulates haematopoietic stem cell fate. Nat Commun. 2015;6:7901. doi:10.1038/ncomms8901
90. Li P, Gao L, Cui T, et al. Cops5 safeguards genomic stability of embryonic stem cells through regulating cellular metabolism and DNA repair. Proc Natl Acad Sci U S A. 2020;117(5):2519–2525. doi:10.1073/pnas.1915079117
91. Bahat A, Goldman A, Zaltsman Y, et al. MTCH2-mediated mitochondrial fusion drives exit from naïve pluripotency in embryonic stem cells. Nat Commun. 2018;9(1):5132. doi:10.1038/s41467-018-07519-w
92. Goldman A, Mullokandov M, Zaltsman Y, et al. MTCH2 cooperates with MFN2 and lysophosphatidic acid synthesis to sustain mitochondrial fusion. EMBO Rep. 2024;25(1):45–67. doi:10.1038/s44319-023-00009-1
93. Zhou X, Zhang K, He Z, et al. Downregulated miR-150 in bone marrow mesenchymal stem cells attenuates the apoptosis of LPS-stimulated RAW264.7 via MTCH2-dependent mitochondria transfer. Biochem Biophys Res Commun. 2020;526(3):560–567. doi:10.1016/j.bbrc.2020.03.098
94. Goldman A, Rodríguez-Casuriaga R, González-López E, et al. MTCH2 is differentially expressed in rat testis and mainly related to apoptosis of spermatocytes. Cell Tissue Res. 2015;361(3):869–883. doi:10.1007/s00441-015-2163-2
95. Wu SX, Wan RD, Wang GW, et al. Comparative proteomic analysis identifies differentially expressed proteins associated with meiotic arrest in cattle-yak hybrids. Proteomics. 2023;23(12):e2300107. doi:10.1002/pmic.202300107
96. Zhou G, Liu J, Li X, et al. Silica nanoparticles inducing the apoptosis via microRNA-450b-3p targeting MTCH2 in mice and spermatocyte cell. Environ Pollut. 2021;277:116771. doi:10.1016/j.envpol.2021.116771
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.