")
Back to Journals » Journal of Inflammation Research » Volume 17
Recurrent Coronary Vasospasm in a 50-Year-Old Woman with Granulomatous Polyangiitis: A Case Report
Authors Hou L, Zhao J, He T, Luo Y, Su K, Li Y, Zhu R
Received 6 May 2024
Accepted for publication 2 August 2024
Published 12 August 2024 Volume 2024:17 Pages 5285—5291
DOI https://doi.org/10.2147/JIR.S472889
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Ling Hou,1,* Jinbo Zhao,2,* Ting He,1,* Yinhua Luo,3 Ke Su,2 Yuanhong Li,2 Ruiyang Zhu3
1Central Hospital of Tujia and Miao Autonomous Prefecture, Hubei University of Medicine, Shiyan, People’s Republic of China; 2Cardiovascular Disease Center, Central Hospital of Tujia and Miao Autonomous Prefecture, Hubei University of Medicine, Enshi, Hubei Province, People’s Republic of China; 3Center for Gene Diagnosis and Department of Clinical Laboratory Medicine, Zhongnan Hospital of Wuhan University, Wuhan, Hubei Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yuanhong Li, Cardiovascular Disease Center, Central Hospital of Tujia and Miao Autonomous Prefecture, Hubei University of Medicine, Enshi, 445400, People’s Republic of China, Email [email protected] Ruiyang Zhu, Center for Gene Diagnosis and Department of Clinical Laboratory Medicine, Zhongnan Hospital of Wuhan University, Wuhan, 430071, People’s Republic of China, Email [email protected]
Abstract: Granulomatosis with polyangiitis (GPA) is a necrotizing granulomatous vasculitis classified as an autoimmune small-vessel vasculitis. Clinically, approximately 80% of affected organs in GPA involve the upper/lower respiratory tract and kidneys, with cardiovascular system involvement being rare. Here, we report a case of a 50-year-old female patient who presented with sudden-onset chest pain lasting for 1 hour. The patient had normal body temperature, and markers of infection such as C-reactive protein and erythrocyte sedimentation rate were within normal limits. Electrocardiography revealed ST-segment elevation in inferior, precordial, and posterior leads. Emergency coronary angiography showed no significant obstructive disease, prompting consideration of vasospastic angina given the patient’s recurrent chest pain symptoms and findings on laboratory and imaging studies. The patient underwent treatment including coronary vasospasm antagonists and immunomodulation, resulting in clinical improvement and subsequent discharge. During a 7-month follow-up period, the patient did not experience any further adverse cardiovascular events.
Keywords: granulomatosis with polyangiitis, vasospastic angina, case report
Introduction
Granulomatosis with polyangiitis, also known as Wegener’s granulomatosis, is a necrotizing vasculitis that affects the ears, nose, and upper and lower respiratory tract. It primarily targets small to medium vessels and has the potential to affect vital organs.1 In clinical presentation, 80% of patients exhibit respiratory tract, kidney, mucous membrane, skin, eye, and nervous system involvement.2 GPA is linked to anti-neutrophil cytoplasmic antibodies (ANCA) and typically manifests as cytoplasmic ANCA positivity (C-ANCA) or anti-proteinase 3 (anti-PR3) antibody positivity.3 Additionally, GPA can impact the heart, cardiovascular manifestations in GPA may include pericarditis, myocarditis, coronary artery disease, and less frequently, coronary vasospasm.4,5 The prevalence of cardiovascular involvement in GPA varies, with some studies reporting it in up to 30–50% of patients. Coronary vasospasm, however, remains an exceptionally rare presentation. Here, we report a case of vasospastic angina triggered by granulomatosis with polyangiitis for the first time, showcasing various aspects of this systemic disease, such as a thorough clinical history, specialized knowledge comprehension, engagement of a multidisciplinary team (MDT), and the critical nature of prompt intervention during severe symptoms.
Case Presentation
We report the case of a 50-year-old woman transferred to our hospital from an external facility due to acute-onset chest pain at 23:00. The electrocardiogram (ECG) conducted at the external facility displayed ST-segment elevation in the inferior, anterolateral, and posterior leads (refer to Figure 1), prompting the initiation of dual antiplatelet therapy (aspirin + ticagrelor). The patient had a history of bilateral peroneal neuropathy of unknown origin over several years and a cerebral hemorrhage 6 years prior, necessitating decompressive craniotomy. The patient is 160 cm tall and weighs 58 kg. She had well-managed hypertension (Long-term oral administration of diltiazem tablets) with no family history of coronary artery disease, and lacked traditional risk factors such as alcohol use, smoking, diabetes, or dyslipidemia. The case report was approved by the patient through the signed informed consent form and by the Enshi Tujia and Miao Autonomous Prefecture Central Hospital hospital’s ethics committee, thus meeting the ethical review standards.
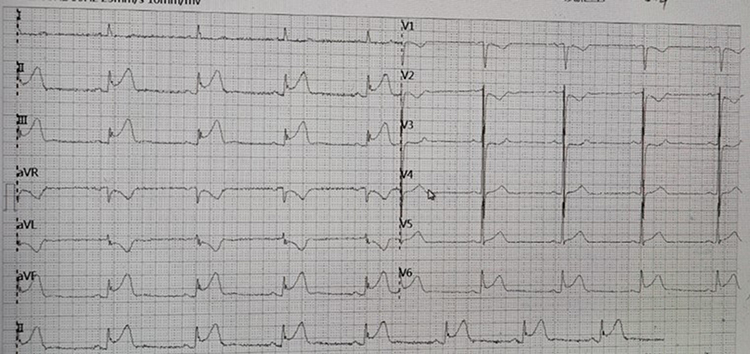 |
Figure 1 The patient’s electrocardiogram from an external hospital shows ST-segment elevation in the inferior wall, anterior lateral wall, and posterior wall leads. |
Physical examination, including cardiovascular evaluation, revealed no notable findings upon admission. Subsequent ECG indicated persistent ST-segment elevation in the inferior, anterolateral, and posterior leads (see Figure 2). The patient continued to experience severe retrosternal squeezing pain combined with dyspnea. Laboratory assessments unveiled elevated levels of troponin T (50 ng/l) and myoglobin (89 ng/L). The dynamic follow-up of biomarkers after hospitalization is depicted in Figure 3. Complete blood count, liver and renal function tests, and lipid profile results were all within normal ranges, while inflammatory markers (C-reactive protein, erythrocyte sedimentation rate) returned negative. Bedside echocardiography demonstrated a normal left ventricular ejection fraction (64%) with segmental wall motion irregularities in the inferior and posterior walls.
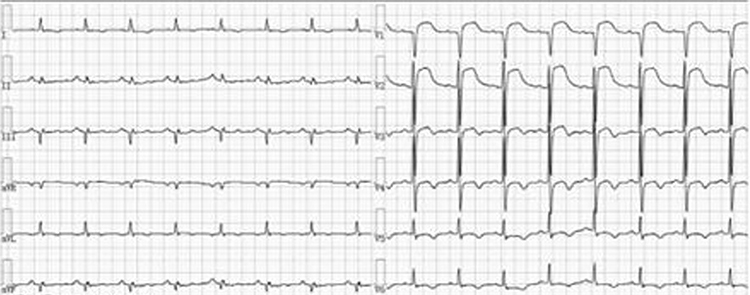 |
Figure 2 The pre-PCI electrocardiogram of the patient shows ST-segment elevation in the inferior wall, anterior lateral wall, and posterior wall leads, with no significant change compared to the electrocardiogram from the external hospital. |
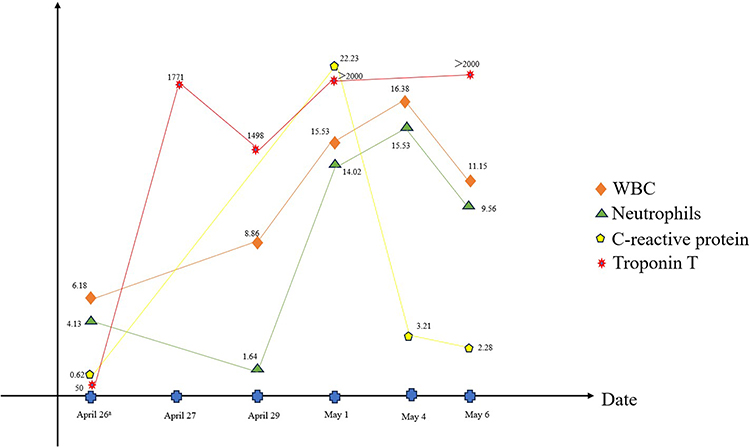 |
Figure 3 Dynamic follow-up of biomarkers. |
The patient’s condition rapidly deteriorated upon admission, culminating in a diagnosis of acute inferior myocardial infarction based on clinical symptoms and auxiliary tests. Emergency coronary angiography, as shown in Figure 4, displayed normal findings. The patient’s angina was alleviated through intracoronary administration of nitrates. With no hemodynamic abnormalities detected in the angiography and the relief of chest pain symptoms by calcium channel blockers, along with an ST segment elevation ≥0.1 mV on the ECG, it suggests that the patient’s angina symptoms could be attributed to coronary artery spasm. Following the procedure, symptomatic management involving continuous nicorandil infusion and antiplatelet therapy was initiated, leading to hemodynamic improvement. Serial troponin I levels decreased to 1498 ng/l.
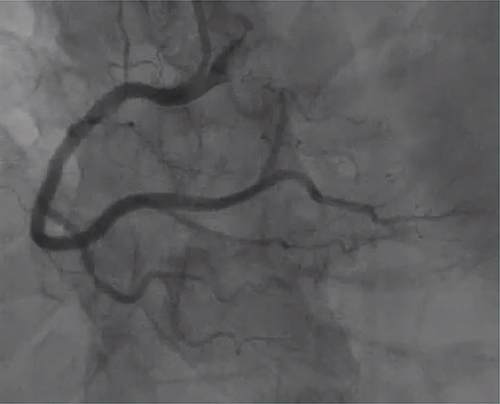 |
Figure 4 Coronary angiography shows non-obstructive coronary arteries. |
Nevertheless, the patient encountered recurrent chest pain progressing to cardiogenic shock 3 days later, with troponin T levels exceeding 2000 ng/l. The patient underwent tracheal intubation, mechanical ventilation, and received vasopressor support for prompt resuscitation. While under mechanical ventilation, sedation was maintained through the administration of sedatives. Subsequently, her chest pain escalated in frequency. Further imaging studies, including PET-CT and myocardial perfusion scintigraphy, were carried out to investigate the etiology of recurrent coronary vasospasm, revealing increased myocardial metabolism in the left ventricular septum, lateral wall, and inferoapical region indicative of ischemia (Figures 5 and 6). Myocarditis-induced coronary vasospasm was initially suspected, although the patient did not exhibit recent symptoms of viral infection and was thoroughly evaluated to rule out viral myocarditis, including virological examination, as well as other infections like tuberculosis. In addition, systemic Lupus Erythematosus (SLE) was considered in the differential diagnosis due to overlapping clinical features such as vasculitis and systemic symptoms. However, the absence of specific diagnostic criteria for SLE, including a negative anti-nuclear antibody (ANA) test and the lack of clinical manifestations typically associated with SLE, such as malar rash and photosensitivity, led to its exclusion. Consequently, the definitive diagnosis established was GPA.
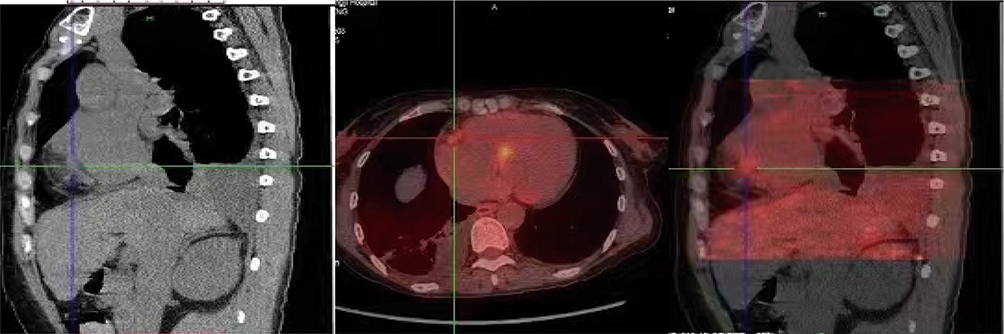 |
Figure 5 Cardiac PET-CT suggests: diffuse elevated myocardial metabolism in the interventricular septum, lateral wall, and inferior wall-apical myocardium; patchy mild metabolism in the right ventricular epicardial region; increased metabolism in the proximal segment of the right coronary artery; mild increase in metabolism in the distal segment of the left circumflex artery, suggestive of myocardial inflammatory changes. |
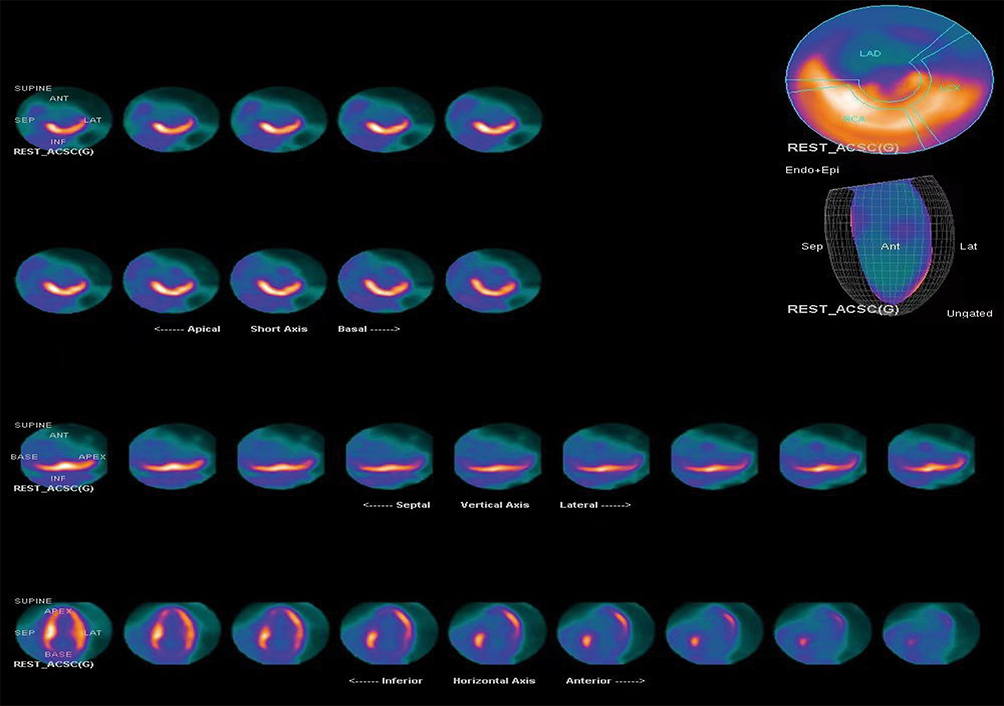 |
Figure 6 Myocardial nuclear imaging reveals perfusion-metabolism mismatch in the apical and middle-inferior walls of the heart. |
During the assessment of the coronary vasospasm etiology, the patient complained of escalating right lower limb pain since admission. Despite analgesic treatment, her symptoms persisted. Electromyography of the lower limbs confirmed bilateral peroneal nerve damage. A neurology consultation was sought, and comprehensive testing for ANCA and anti-nuclear antibodies spectrum revealed positivity for anti-neutrophil cytoplasmic antibodies targeting proteinase 3. Granulomatosis with polyangiitis-induced vasospastic angina was considered. Due to the patient’s frail health, she declined biopsy. Following diagnosis and literature review, treatment with diltiazem + glucocorticoids + immunoglobulin modulation therapy was initiated, resulting in decreased frequency of chest pain attacks and alleviation of right lower limb pain. The patient was discharged on the 14th day with a favorable outcome and instructed to maintain oral prednisolone therapy. Over the subsequent 7 months, she did not experience any further adverse cardiovascular events. We confirm that written consent has been obtained from the patients mentioned in this case report for the publication of their details and accompanying images. The consent form is available for review upon request.
Discussion
GPA formerly known as Wegener’s granulomatosis, is an autoimmune vasculitis categorized as antineutrophil cytoplasmic antibody-associated vasculitis (AAV), which also encompasses eosinophilic granulomatosis with polyangiitis (EGPA) and microscopic polyangiitis (MPA).6 GPA, the most prevalent subtype of AAV, is identified by the presence of c-ANCA and antibodies against proteinase 3, leading to necrotizing granulomatous vasculitis mainly affecting medium to small-sized vessels.7 GPA is relatively uncommon, with an incidence of 5 cases per 100,000 individuals in Europe, higher in Northern Europe, and predominantly affecting Caucasians, showing no gender bias, with an average onset age of about 40 years.8 The etiology of GPA remains unclear, although genetic predisposition, environmental factors, microbial infections, medications, novel coronavirus infection, vaccination, and hematopoietic stem cell transplantation are suggested as contributing factors.9 The pathogenesis of GPA is associated with deficiencies in innate and adaptive immunity, B-cell dysfunction, and the pathogenic production of antineutrophil cytoplasmic antibodies (ANCAs).10
Our research identifies the first documented case in medical literature of GPA causing coronary artery vasospastic angina. Previous research indicates that 76% of GPA cases are positive for C-ANCA antibodies, 82.2% for PR-3 ANCA antibodies, 13% for P-ANCA antibodies, and 8.1% for MPO-ANCA antibodies. In severe GPA affecting multiple systems, the rates of positivity for C-ANCA and PR-3 ANCA rise to 78% and 76%, respectively.11,12 GPA can impact various organ systems, such as the skin, joints, pericardium, heart, eyes, and peripheral nervous system.13 Further investigation of the patient’s medical history revealed initial peripheral nervous system involvement leading to a multidisciplinary consultation. Laboratory tests confirmed PR-3 positivity, resulting in a GPA diagnosis.
Prior to the introduction of glucocorticoids and other immunosuppressive agents, the prognosis for GPA was bleak, with untreated systemic cases often resulting in fatality.14 Currently, a treatment regimen combining glucocorticoids and cyclophosphamide has achieved remission in over 80% of patients, significantly reducing the five-year mortality rate to 10–15%.15 Recent research suggests the utility of intravenous immunoglobulin therapy for refractory or relapsing GPA cases.16 Due to our patient’s recurrent episodes of coronary artery vasospasm, we opted for a treatment approach incorporating glucocorticoids and immunoglobulins. Subsequent monitoring confirmed the treatment’s effectiveness.
Our patient had a history of neuropathy and presented with chest pain, displaying electrocardiographic evidence indicative of ST-segment elevation myocardial infarction. However, coronary angiography revealed no significant stenosis, while PET-CT and myocardial perfusion scintigraphy exhibited diffuse myocardial hypermetabolism. Administration of calcium channel blockers alleviated the patient’s chest pain episodes. Following comprehensive consultations and diagnostic medication, the patient was ultimately diagnosed with coronary vasospastic angina. Cardiac involvement in GPA is infrequent, characterized by manifestations like pericarditis, myocarditis, valvular issues, conduction abnormalities, myocardial infarction, and heart failure.17–21 Research indicates that cardiovascular involvement in ANCA-associated vasculitis is a harbinger of poor prognosis and increased mortality rates.22,23
Certainly, our case report has several limitations. Firstly, we did not utilize IVUS (intravascular ultrasound) or OCT (optical coherence tomography), which restricted a detailed assessment of coronary artery lesions. Additionally, we did not perform ergonovine or acetylcholine provocation tests, which are crucial for evaluating coronary vasospasm. The absence of these diagnostic methods may have affected the accuracy of our diagnosis of coronary vasospasm. Moreover, the study is based on a single case, limiting the generalizability of the findings and lacking long-term follow-up data. As a result, our observations suggest a potential association rather than a definitive causal relationship. Future research should involve a larger number of cases, diverse diagnostic tools, and extended follow-up to better validate and understand the complexities in this area.
Our case study sheds light on a rare occurrence where granulomatosis with polyangiitis (GPA) can trigger angina through coronary artery spasm, underscoring the significance of a thorough evaluation of GPA’s systemic effects. This discovery not only broadens the array of clinical presentations associated with GPA but also advises healthcare providers to contemplate GPA as a potential diagnosis in cases of analogous symptoms, facilitating timely and precise diagnosis and intervention.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kronbichler A, Bajema IM, Bruchfeld A. et al. Diagnosis and management of ANCA-associated vasculitis. Lancet. 2024;403(10427):683–698. doi:10.1016/S0140-6736(23)01736-1
2. Greco A, Marinelli C, Fusconi M, et al. Clinic manifestations in granulomatosis with polyangiitis. Int J Immunopathol Pharmacol. 2016;29(2):151–159. doi:10.1177/0394632015617063
3. Sharma A, Dogra S, Sharma K. Granulomatous Vasculitis. Dermatol Clin. 2015;33(3):475–487. doi:10.1016/j.det.2015.03.012
4. Czaplińska M, Dorniak K, Lizakowski S, et al. Cardiac involvement as a fatal complication of granulomatosis with polyangiitis. Pol Arch Intern Med. 2021;131(1):73–74. doi:10.20452/pamw.15730
5. Parmar MK, Alikhan M, Hsu VM, et al. Echocardiogram: the GPS to GPA’s Heart (Granulomatosis with Polyangiitis). Case Rep Rheumatol. 2019;2019:7609386. doi:10.1155/2019/7609386
6. Müller A, Krause B, Kerstein-Stähle A, et al. Granulomatous Inflammation in ANCA-Associated Vasculitis. Int J Mol Sci. 2021;22(12):6474. doi:10.3390/ijms22126474
7. Marquez J, Flores D, Candia L, et al. Granulomatous vasculitis. Curr Rheumatol Rep. 2003;5(2):128–135. doi:10.1007/s11926-003-0040-6
8. Lutalo PM, D’Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis). J Autoimmun. 2014;48-49:94–98. doi:10.1016/j.jaut.2014.01.028
9. Brieske C, Lamprecht P, Kerstein-Staehle A. Immunogenic cell death as driver of autoimmunity in granulomatosis with polyangiitis. Front Immunol. 2022;13:1007092. doi:10.3389/fimmu.2022.1007092
10. Muller K, Lin JH. Orbital granulomatosis with polyangiitis (Wegener granulomatosis): clinical and pathologic findings. Arch Pathol Lab Med. 2014;138(8):1110–1114. doi:10.5858/arpa.2013-0006-RS
11. Finkielman JD, Lee AS, Hummel AM, et al. ANCA are detectable in nearly all patients with active severe Wegener’s granulomatosis. Am J Med. 2007;120(7):
12. Chen JW, Zhan JY, Liang SP, et al. A patient with refractory proliferative lupus nephritis treated with telitacicept: a case report. Int J Rheum Dis. 2023;26(7):1417–1421. doi:10.1111/1756-185X.14752
13. Binda V, Moroni G, Messa P. ANCA-associated vasculitis with renal involvement. J Nephrol. 2018;31(2):197–208. doi:10.1007/s40620-017-0412-z
14. Emmi G, Bettiol A, Gelain E, et al. Evidence-Based Guideline for the diagnosis and management of eosinophilic granulomatosis with polyangiitis. Nat Rev Rheumatol. 2023;19(6):378–393. doi:10.1038/s41584-023-00958-w
15. Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363(3):221–232. doi:10.1056/NEJMoa0909905
16. Álamo B S, Moi L, Bajema I, et al. Long-term outcomes and prognostic factors for survival of patients with ANCA-associated vasculitis. Nephrol Dial Transplant. 2023;38(7):1655–1665. doi:10.1093/ndt/gfac320
17. Gałąska R, Kulawiak-Gałąska D, Czuszyńska Z, et al. A rare case of complex cardiac involvement in granulomatosis with polyangiitis. Pol Arch Intern Med. 2017;127(1):63–65. doi:10.20452/pamw.3914
18. Colin GC, Vancraeynest D, Hoton D, et al. Complete heart block caused by diffuse pseudotumoral cardiac involvement in granulomatosis with polyangiitis. Circulation. 2015;132(17):e207–210. doi:10.1161/CIRCULATIONAHA.115.017843
19. Suleymenlar G, Sarikaya M, Sari R, et al. Complete heart block in a patient with Wegener’s granulomatosis in remission--A case report. Angiology. 2002;53(3):337–340. doi:10.1177/000331970205300312
20. Santos LPS, Bomfim VG, Bezerra CF, et al. Heart conduction system defects and sustained ventricular tachycardia complications in a patient with granulomatosis with polyangiitis. A case report and literature review. Rev Bras Ter Intensiva. 2017;29(3):386–390. doi:10.5935/0103-507X.20170052
21. Forstot JZ, Overlie PA, Neufeld GK, et al. Cardiac complications of Wegener granulomatosis: a case report of complete heart block and review of the literature. Semin Arthritis Rheum. 1980;10(2):148–154. doi:10.1016/0049-0172(80)90005-0
22. Pakbaz M, Pakbaz M. Cardiac Involvement in Eosinophilic Granulomatosis with Polyangiitis: a Meta-Analysis of 62 Case Reports. J Tehran Heart Cent. 2020;15(1):18–26.
23. Hazebroek MR, Kemna MJ, Schalla S, et al. Prevalence and prognostic relevance of cardiac involvement in ANCA-associated vasculitis: eosinophilic granulomatosis with polyangiitis and granulomatosis with polyangiitis. Int J Cardiol. 2015;199:170–179. doi:10.1016/j.ijcard.2015.06.087
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.