")
Back to Journals » Vascular Health and Risk Management » Volume 20
Repurposing Metformin for the Treatment of Atrial Fibrillation: Current Insights
Authors Sarkar A , Fanous KI , Marei I, Ding H, Ladjimi M, MacDonald R , Hollenberg MD , Anderson TJ, Hill MA, Triggle CR
Received 12 February 2024
Accepted for publication 5 June 2024
Published 21 June 2024 Volume 2024:20 Pages 255—288
DOI https://doi.org/10.2147/VHRM.S391808
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Daniel Duprez
Aparajita Sarkar,1 Kareem Imad Fanous,1 Isra Marei,2 Hong Ding,2 Moncef Ladjimi,3 Ross MacDonald,4 Morley D Hollenberg,5 Todd J Anderson,6 Michael A Hill,7 Chris R Triggle2
1Department of Medical Education, Weill Cornell Medicine-Qatar, Doha, Qatar; 2Department of Pharmacology & Medical Education, Weill Cornell Medicine- Qatar, Doha, Qatar; 3Department of Biochemistry & Medical Education, Weill Cornell Medicine-Qatar, Doha, Qatar; 4Health Sciences Library, Weill Cornell Medicine-Qatar, Doha, Qatar; 5Department of Physiology & Pharmacology, and Department of Medicine, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada; 6Department of Cardiac Sciences and Libin Cardiovascular Institute, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada; 7Dalton Cardiovascular Research Center & Department of Medical Pharmacology & Physiology, School of Medicine, University of Missouri, Columbia, Missouri, USA
Correspondence: Aparajita Sarkar; Chris R Triggle, Email [email protected]; [email protected]
Abstract: Metformin is an orally effective anti-hyperglycemic drug that despite being introduced over 60 years ago is still utilized by an estimated 120 to 150 million people worldwide for the treatment of type 2 diabetes (T2D). Metformin is used off-label for the treatment of polycystic ovary syndrome (PCOS) and for pre-diabetes and weight loss. Metformin is a safe, inexpensive drug with side effects mostly limited to gastrointestinal issues. Prospective clinical data from the United Kingdom Prospective Diabetes Study (UKPDS), completed in 1998, demonstrated that metformin not only has excellent therapeutic efficacy as an anti-diabetes drug but also that good glycemic control reduced the risk of micro- and macro-vascular complications, especially in obese patients and thereby reduced the risk of diabetes-associated cardiovascular disease (CVD). Based on a long history of clinical use and an excellent safety record metformin has been investigated to be repurposed for numerous other diseases including as an anti-aging agent, Alzheimer’s disease and other dementias, cancer, COVID-19 and also atrial fibrillation (AF). AF is the most frequently diagnosed cardiac arrythmia and its prevalence is increasing globally as the population ages. The argument for repurposing metformin for AF is based on a combination of retrospective clinical data and in vivo and in vitro pre-clinical laboratory studies. In this review, we critically evaluate the evidence that metformin has cardioprotective actions and assess whether the clinical and pre-clinical evidence support the use of metformin to reduce the risk and treat AF.
Keywords: metformin, atrial fibrillation, AMPK, hyperglycemia, hypoglycemia, cardiac metabolism, cardiovascular protection, atrial remodeling
Graphical Abstract:
Introduction
Cardiovascular diseases (CVD) are a major global concern due to an almost two-fold increase in prevalence in the past three decades, which in 2019 saw 523 million new cases and accounted for 32% of all deaths.1,2. Although there has been an overall decline in mortality rates due to CVD, albeit less so in developing countries, there is still the need to improve treatment and survival.3 There have been many new additions to the therapeutic armamentarium to treat CVD; however, these new drugs are not necessarily universally cost-effective. A study in 2003 by Garattini & Bertele4 concluded that the most recently approved cardiovascular agents in Europe for the treatment of hypertension, arrhythmias, and thrombosis have only added minimally to progress in this field and, furthermore, that the newer drugs are more expensive than similar existing drugs in terms of efficacy and safety, and provide questionable economic or medical benefit. Twenty-one years later in 2024 this conclusion could certainly be considered as premature due to the Introduction of what have been described in terms of cardiovascular benefits “game changers”, namely glucagon-like peptide receptor agonists (GLP-1 RAs) with exenatide approved by the FDA in 2005 (and for semaglutide in late 2017), and sodium-glucose co-transporter-2 (SGLT-2) inhibitors, canagliflozin approved in 2013 for the treatment of type 2 diabetes (T2D).5–7 Regardless, the question remains: “Can already approved, and usually less expensive, drugs, be evaluated for new indications”? This review addresses this question and specifically critically analyses the potential use of metformin to reduce the risk of AF.
Affordability remains an important factor in determining the effectiveness of medical interventions to treat some diseases, such as CVD (including AF), diabetes and obesity, as the cost of newer drugs may be beyond the budgets of a large percentage of the global population and particularly those residing in low- and middle-income countries. Income inequalities may also result in less access to quality and equitable healthcare and programmes for prevention, early detection, and treatment of CVD. For instance, it has been estimated that more than 25% of the global population has minimal or no access to appropriate drug therapies.8 Consequently, detection and diagnosis are often late and appropriate treatment is delayed, resulting in premature mortality in these patients, which for CVD results is 75%.9 At an individual level, the high cost of appropriate medications contributes to increasing poverty, and at the national level, a substantial burden on the overall economy of the country.2
A potential solution to the high cost of newly approved drugs lies in repurposing old, existing and more affordable drugs to treat CVD, a concept frequently referred to as “Old Drugs for New Tricks”. However, a need remains to conduct appropriate clinical trials for existing drugs for the new indications and conditions.10 An example of such a drug is the biguanide N,N-dimethylbiguanide, which is best known as metformin, has a long history of use in the treatment of T2D, and in recent years has been extensively investigated for the treatment of a number of other conditions, including aging, cancer and more recently, COVID-19.11–13 Based on the success rates for repurposing drugs there is merit to this process, as it has been reported that approval for repurposing a drug has a success rate of 30% versus 11% for a newly developed drug.14
Diabetes is a well-recognised risk factor for the development of CVD15 and is also a risk factor for AF in both males and females; the risk and burden of AF (that is the period of time in AF) is also positively correlated with higher-glycated haemoglobin (HbA1C) levels.16–22 A meta-analysis of data from 29 studies involving over 8 million patients concluded that diabetes increased the risk of AF by 48%, with women at greater risk.23 Importantly, treatment with the anti-diabetes drug metformin has been reported to provide therapeutic benefits for patients with cardiac and vascular diseases, including AF.24–38 However, much of this evidence is based on clinical data from observational studies and confirmation is required from randomised placebo-controlled prospective studies.28 Furthermore, it has not been clearly established whether metformin has CVD protective benefits in patients without diabetes because existing data are controversial, as evident from the Carotid Atherosclerosis: MEtformin for insulin ResistAnce (CAMERA) study.39
A Scopus analysis (Figure 1) indicates the increasing interest in metformin and its potential for the treatment of atrial arrhythmias and AF.
In order, to critically review protocol design and the pre-clinical and clinical data that support the potential use of metformin to treat AF it is important to understand the pharmacokinetic and pharmacodynamic properties of metformin and the controversies over the cellular properties of the drug. AF is the most frequently reported sustained cardiac arrhythmia, accounting for a third of all arrhythmia-related hospitalizations and its prevalence, currently 0.4% to 1% of the general population, is increasing as the population ages with estimates that numbers in the USA will exceed 10 million by 2050.40,41
The putative benefits of metformin as a drug for risk reduction in CVD, including AF, may entirely be secondary to its therapeutic efficacy in the setting of diabetes, as an anti-diabetes drug, thus raising the question as to whether metformin, and other anti-diabetes drugs, would also reduce CVD risk in subjects without diabetes? Of interest, in 2023 the American Diabetes Association revised their recommendations for the use of metformin and T2D and for those patients with pre-existing atherosclerotic CVD risk factors, including heart failure and chronic kidney disease, where the use of gliflozins and GLP-1 RAs is now recommended (see: https://doi.org/10.2337/cd23-as01). However, there are disadvantages to using gliflozins and GLP-1 RAs as adjuncts to treat AF that include cost-effectiveness per patient, which may also affect global accessibility. Thus, there is value in considering metformin as an adjunct for AF given the extensive database on metformin’s clinical use since the 1960s.
Metformin
Metformin is a synthetic biguanide with a long history of clinical use based on the knowledge that naturally occurring guanidines, notably galegine, found in French Lilac (‘Galega Officinalis’), lowered blood glucose levels.13,38,42–46 Metformin is a first-line drug approved for the treatment of T2D. Commonly prescribed as GlucophageR, it has over 60 years of history in the treatment of T2D. Although metformin was approved and in use in Europe from the 1960s for the treatment of T2D, its use in the USA was not approved by the FDA until 1994. Approximately 120 to 150 million people globally use metformin, with the vast majority using it for the treatment of T2D.13,47 Metformin is a well-researched drug, and an extensive literature including many review articles support its benefits, and lack of serious side effects, emphasizing its longstanding importance in the treatment of T2D.13,47–54 In addition to its demonstrated safety, metformin is inexpensive and is effective for the vast majority of patients with T2D.50 Metformin also has several established off-label uses such as treating pre-diabetes to prevent the development of overt T2D, for weight-loss, and for the treatment of polycystic ovary syndrome (PCOS) see section 1.1.3.13,54,55 The use of metformin has also been investigated as an anti-aging drug, for Alzheimer’s Disease and other dementias, as well as for reducing the risk of developing certain forms of cancer. Metformin has also been evaluated for the treatment of SARS-CoV-2 infection, thereby pointing to a wide therapeutic potential of repurposing metformin for multiple diseases.12,13,46,54,56–62
It is important to stress that the evidence supporting the therapeutic efficacy of metformin as ‘a drug for all reasons’ is highly controversial and has been vigorously and critically evaluated, particularly with respect to the experimental design and data interpretation of in vitro studies as well as to conclusions based on observational clinical studies.12,13 The results of such studies have not always been positive as has been the case for the use of metformin as an adjunct for the treatment of breast cancer where data from randomized control studies of metformin as an adjunct for the prevention and treatment of cancer have not been supportive.63,64 The reason for the differences in interpretation can be linked to time-related biases in the analysis of the data from observational studies that bias interpretation of the data towards a positive effect of the drug, when in fact no benefit has occurred and this issue also is evident in studies of metformin and AF.65–68 In addition, because the predominance of data is derived from patients with T2D who have been treated with metformin, it is difficult to separate benefits from the therapeutic actions of metformin as an anti-diabetes drug with its anti-hyperglycemic and insulin-sensitizing actions from unrelated and distinct putative pleiotropic effects. However, some evidence for the latter does exist including data based on an analysis of over 40,000 patients with T2D that showed that patients who terminated treatment with metformin, but did not experience increases in HbA1c levels or insulin use, were at greater risk of developing dementia therefore inferring that protection was at least in part independent of metformins’ action as an anti-diabetes drug.62 Nevertheless, there remains the need for data obtained from prospective studies comparing patients without diabetes to those with diabetes. Collectively, these and other concerns should be considered before recommending the repurposing of a drug, even for metformin, which has relatively few potential serious side effects.69,70
Pharmacokinetics of Metformin (Route of Administration, Absorption, Distribution, Metabolism, Elimination)
A comprehensive understanding of the pharmacokinetic properties of metformin is not only important for optimizing the clinical use of metformin but also for the appropriate design and interpretation of pre-clinical studies that involve in vivo studies in animals as well as in vitro cell-based studies. Metformin is a basic hydrophilic drug that in humans, and as far as is known other species, is not metabolized and is excreted via the urine unchanged; however, recent data indicate that some bacteria associated with waste water have developed the ability to utilise metformin as a carbon source with guanylurea as an intermediate metabolite.71,72 Metformin has a pKa of approximately 11.5 and exists predominantly as a cation at physiological pH. It has a plasma half-life that has been reported to range from 1.5 to 8 hours, and its mean rate of renal clearance is 510 ± 120 mL/min.43,73–77 Table 1 summarizes the pharmacokinetic properties of metformin.
 |
Table 1 Pharmacokinetic Properties of Metformin |
Pharmacodynamics and Mechanism of Action as an Anti-Hyperglycemic Drug
Metformin reduces basal and postprandial blood glucose levels by increasing insulin sensitivity while also decreasing hepatic glucose production and reducing intestinal glucose absorption.45,54 Despite intensive investigation, the mechanism by which metformin provides its exact anti-hyperglycemic effect(s) remain unclear, although the preponderance of data indicates that the primary target involves the activation of AMP-activated protein kinase (AMPK).84 Although it remains controversial as to the specific pathway(s) which are responsible for the activation of AMPK via phosphorylation of the AMPK α catalytic subunit at Thr-172, studies suggest the involvement of other kinases such as the serine threonine kinase liver kinase B1 and (LKB1/STK11), which is also a tumour suppressor, and the DNA-damage sensor, Ataxia Telangiectasia Mutated (ATM) gene.77,85–88 See Figure 2 for details. The putative roles of LKB1 and ATM (ATM encodes for phosphatidylinositol 3/phosphatidylinositol 4 (PI3/PI4)-kinases) as targets for metformin are of particular interest because of the epidemiological and, particularly, pre-clinical data suggesting that the use of metformin reduces the risk and development of several types of cancer and potentially also involved in other beneficial effects of metformin including CVD.11,13,89–94 Other putative sites of action for metformin include the NAD-dependent deacetylase, sirtuin-1, that is known to positively regulate endothelial function,91,92 and the activation of LKB1 and AMPK via the orphan nuclear receptor, NR4A1, and these actions are reported to contribute to the endothelial-vascular protective action of metformin that potentially are involved in reducing the risk of AF.13,93,94
 |
Figure 1 Papers published for the period 1974–2023 that include terms for metformin and atrial fibrillation. Note: Created with BioRender.com. |
 |
Figure 2 Putative pathways for activation of AMPK by metformin. Notes: Putative mechanisms for the activation of adenosine monophosphate-activated protein kinase (AMPK) by metformin. One pathway is via the inhibition of mitochondrial complex I of the electron transport chain (see El-Mir et al 2000; Owen et al 200095,96). Inhibition of complex 1 results in an increase in the ratio of adenosine monophosphate (AMP) and adenosine diphosphate (ADP) to adenosine triphosphate (ATP), which in turn results in the activation of AMPK. Some studies indicate a role for the upstream candidate serine/threonine kinase 11 (STK11) (also known as liver kinase B1 (LKB1)). Metformin effects via LKB1, a tumor suppressor, and the DNA-damage sensor, ATM (Ataxia Telangiectasia Mutated gene), which encodes for phosphatidylinositol 3/phosphatidylinositol 4 (PI3/PI4)-kinases, may contribute to the putative beneficial effects of metformin to reduce the risk of a number of cancers.11,77,85–90 Another alternative pathway is activation of AMPK via a mechanism dependent on the expression of the Nuclear Receptor Subfamily 4 Group A Member 1, NR4A1 (also referred to Nur77), and subsequent activation of the STK11/AMPK signaling cascade.94 Created with BioRender.com. |
Metformin has been reported to have LKB1- and AMPK-independent actions that result in the inhibition of hepatic glucose production as has been shown in AMPK knockout mice lacking the expression of AMPK where the effects of metformin appear to be preserved.48 Based primarily on data from in vitro studies, metformin has been shown at high concentrations (mM) relative to those obtained in vivo (low micromolar) to inhibit mitochondrial complex I, which increases the cellular AMP:ATP ratio in the cell leading to the activation of AMPK.95–97 Bridges et al (2023) have provided evidence using cryo-electron microscopy that a hydrophobic derivative of metformin, IM1092, and a very potent inhibitor of complex 1 with an IC50 of 86 μM, binds to the ubiquinone channel of complex 1.97 However, although inhibition of complex 1 is an attractive hypothesis and can be demonstrated by using mM concentrations of metformin in in vitro studies inhibition of complex 1 does not appear to explain metformin’s therapeutic benefits as when used clinically evidence is lacking that metformin accumulates to sufficiently high concentrations to exert a sustained inhibitory action on mitochondrial complex 1.13,76,98–104
AMPK activation plays a major role in the regulation of glucose and lipid metabolism. Activated AMPK phosphorylates various targets including HMG-CoA reductase, mammalian target for rapamycin (mTOR), acetyl-CoA carboxylase (ACC), ACC-2, glycerol-3-phosphate acyltransferase, and carbohydrate response element-binding protein, leading to inactivation while also suppressing expression of the transcription factor, SREBP-1, that is involved in lipid metabolism and discussed in more detail in Metformin AMPK and Cardiac Metabolism.105,106 AMPK also increases mitochondrial biogenesis by activating sirtuin-1, and increasing the expression of the peroxisome proliferator-activated receptor-gamma coactivator (PGC-1α) in the nucleus.107 Activation of hepatic AMPK increases fatty acid beta-oxidation and mitochondrial biogenesis, while inhibiting triglyceride, fatty acid, cholesterol synthesis and insulin signalling via the nutrient-sensitive complex mTOR and the RAPTOR gene, and thereby decreasing gluconeogenesis, and the dysregulation of AMPK is linked to the development and progression of metabolic syndrome and heart disease, which will result in an increased risk of AF.105 Metformin, albeit at high concentrations (0.5 mM), has been reported to downregulate the expression of the CYP3A4 gene, by disrupting the coactivation of PXR with SRC1 (CYP3A4 plays an important role in the metabolism of a large percentage of drugs).77,108
In addition to its actions on the liver, metformin exerts effects on skeletal muscle via the AMPK pathway that result in increased glucose uptake into myocytes due to increased translocation of GLUT4 transporters to the plasma membrane.77 Metformin also has effects in the GI tract prior to absorption where it can interact with the microbiome, modify GLP-1 levels, and thereby contribute to the anti-hyperglycemic actions of metformin as well as effects on satiety.109
AMPK plays a key role in the regulation of endothelial-vascular smooth muscle function and the regulation of blood flow. AMPK activates recombinant endothelial nitric oxide synthase (eNOS)110 as well as eNOS in mouse and human arterial endothelial cells and cardiomyocytes via the reversible phosphorylation of Ser-1177 resulting in the generation of nitric oxide (NO).110–112 Studies in mice have shown that metformin, by activating the AMPK-eNOS pathway in cardiomyocytes, reduces reperfusion injury post-myocardial infarction and reduces the size of infarcts.113 These data, as reviewed in 2018 by Driver et al, provide supportive evidence for why metformin through activation of AMPK and the downstream effects on metabolic signaling pathways reduces the risk of the development of heart failure, atherosclerosis, coronary artery disease, and chronic kidney disease in patients at high risk for CVD and by inference also AF.114 See Figure 3.
 |
Figure 3 Downstream effects of metformin on metabolism. Notes: Conflicting evidence suggests that metformin activates adenosine monophosphate-activated protein kinase (AMPK) either directly, or indirectly via the upstream serine-threonine liver kinase B1 (LKB1), or via ataxia telangiectasia mutated (ATM). AMPK, sometimes referred to as the “fuel gauge” of the cell, in addition to reducing hepatic gluconeogenesis also induces fatty acid β-oxidation, insulin signalling and mitochondrial biogenesis, and inhibits the synthesis of triglycerides, fatty acids and cholesterol. High levels of metformin may also reduce the expression of CYP3A4. Additionally, via its anti-hyperglycemic actions metformin protects against diabetic cardiomyopathy. In skeletal muscle, AMPK actions result in the increased translocation of Glucose Transporter Type 4 (GLUT4) to the plasma membrane, thereby increasing glucose uptake.105 Created with BioRender.com. |
Further support for the potential benefits for the use of metformin in AF is that hypoglycemia is very rare during therapy with the drug, notably when used as monotherapy, which is why metformin is referred to as a “euglycemic” drug and contrasts with the use of insulins, sulfonylureas, and also GLP-1 RAs if combined with sulfonylureas where the risk of hypoglycemia is a potential serious concern.32,84,115–118
Putative Mechanisms of Action of Metformin and Concerns Over the Interpretation of Data from in vitro Studies
A potentially confounding factor in interpreting the effects of metformin from data derived from in vitro versus in vivo and clinical studies, including those that have focused on AF, relates to the wide concentration and dose ranges that have been employed in such studies.13,101–103,119
The therapeutic dose for metformin in humans ranges from 250 to 2550 mg per day (or maximum 35 mg/kg).45,119 Following absorption from the GI tract, the portal vein concentrations of metformin from animal studies within 1 hour approximate 40 to 70 μM, which rapidly drops to 10–40 μM. However, levels in humans may vary considerably depending on the expression levels of the organic cation transporters (OCT).75,77,79,99,102 As pointed out by Dowling et al,119 the excessively high concentrations and doses that have been used in many of the pre-clinical in vitro and in vivo studies raises concerns over ascribing cellular mechanism(s) of action that explain the drug’s therapeutic benefits. For instance, a number of in vitro studies with metformin have used concentrations that are 25 to 1000 times higher than the plasma concentrations (2–40 μM) that have been reported when metformin is used to treat T2D.119 Similarly for pre-clinical studies where these doses often remain approximately 25 to 45 times higher than the therapeutic range in humans.119 These concentration/dose discrepancies must be carefully assessed when evaluating and comparing data generated from preclinical versus clinical studies and the potential for using metformin to treat AF.13,101–104,119
Off-Label Use of Metformin: Benefits and Concerns for Its Potential Use in AF
Off-label indications of metformin include the treatment of pre-diabetes and thereby reducing the risk for the onset of T2D, treatment of weight gain due to, for instance, antipsychotic drug therapy, and treatment of irregular and inconsistent menstruation due to polycystic ovary syndrome (PCOS) that affects as high as 15% of women of reproductive age and is linked to insulin resistance.55,120,121
Metformin is classified as a Category B drug by the FDA and has generally considered safe for use in pregnancy and has been used off-label in the treatment of gestational diabetes,45 but its use has been questioned in women with both PCOS and gestational diabetes and insulin remains the preferred therapeutic choice in pregnancy.122–125 Furthermore, although no congenital genetic defects have been associated with metformin (but see Wensink et al126), concerns have been raised regarding the effects of metformin on infant weight particularly as relates to low gestational weight gain, and also subsequent obesity in children prenatally exposed to metformin.127–129
Metformin crosses the placenta and can have effects on folate metabolism and may impact growth in the fetus, thus suggesting a need for caution, particularly in the first trimester as placental transfer may vary with genetic polymorphisms in the expression of OCT transporters.130–134 Metformin is also present in breast milk and generally considered insignificant until the dose in the infant crosses 10 mg per kg per day.135 Metformin is approved for use in children above the age of 10, but data on safety for use in children of lower age are not available.136
A concern over the increased use of metformin for pre-diabetes, and by inference to treat AF, in men of reproductive age has come from a 2022 study in Denmark that reported an increase in genital defects in the male offspring,126 supporting concerns raised in a 2018 study on the effects of metformin on the human epigenome137 Further data are required to clarify whether metformin is an endocrine disruptor in humans and whether this may limit the repurposing of metformin for AF and other indications; however, it has been shown that at levels of metformin that are now found in wastewater, and metformin has behavioural and endocrine disruptor effects in zebrafish (Danio rerio) and is an endocrine disruptor in water fleas (Daphnia pulex).138–140 Interestingly, based on data from a metabolomic study, metformin has been detected in plasma samples from patients who have not been prescribed metformin.141
Even though the use of metformin could result in the lowering of insulin dosage based on the results from the REMOVAL trial (REducing with MetfOrmin Vascular Adverse Lesions), metformin is not recommended as an adjunct with insulin for type 1 diabetes.142,143
Metformin: Side Effects and Contraindications, Complications, and Drug Interactions and Potential Impact on Repurposing Metformin for AF
Before promoting the use of metformin for the treatment of AF and other indications, it is important to consider the potential side effects and contraindications that may limit its use. Importantly, such side effects may reduce patient compliance, particularly when chronic use is being considered.
The most frequently reported side effect of metformin is GI distress. Symptoms include nausea, vomiting, upset stomach or diarrhoea, which although not usually serious, may reduce patient compliance in taking the medication.144,145 The GI symptoms are thought to result from the retention of metformin in the enterocytes of the GI tract resulting from saturation of to OCT1-mediated efflux from enterocytes into the interstitial fluid, increased luminal serotonin (due to release via enterochromaffin cells or reduced transport by the serotonin transporter (SERT)), inhibition of intestinal mucosa (mitochondrial complex I), and, or, effects of metformin on the gut microbiome.77,146–148
Chronic use of metformin has also been associated with vitamin B12 deficiency, which potentially can cause megaloblastic anemia and worsen the neuropathy due to pre-existing diabetes.149 Although not fully understood, it is assumed that metformin interferes with the calcium-dependent binding of the cobalt-containing vitamin B12 (also known as cobalamin) to the glycoprotein, “Intrinsic Factor” IF, and thereby prevents cubilin receptor-mediated endocytosis of the vitamin B12.104,150,151 Patients who are prescribed metformin should therefore be monitored periodically and, if appropriate, provided vitamin B12 supplements, especially if the patients have other co-existing conditions such as renal or hepatic impairment.151,152 Metformin-induced vitamin B12 deficiency has been linked to higher frequencies of autonomic neuropathy and linked to a 3.16 fold increase in cardiovascular events, including cardiac arrhythmias, and a 3.17 fold increase in mortality and therefore of importance when considering the chronic use of metformin in subjects with AF.149,153
Metformin is predominantly cleared by the kidneys thereby requiring that the dosage should be adjusted if the patient has reduced renal function. The European Medicines Agency in October 2016 approved metformin to be safe for use in patients with moderately reduced kidney function while remaining contraindicated in those with a glomerular filtration rate (GFR) less than 30 mL/min. Kidney function should be established before use, particularly in geriatric patients. Further, the dosage of metformin should be adjusted in patients on dialysis as metformin does not bind to plasma proteins and because of its small molecular weight is easily dialyzable.45,54 Hypoglycemia with metformin use, although rare, can occur at high levels of metformin (overdose), with reduced renal function, or when used with other anti-diabetes agents or anti-coagulation agents (such as sulfonylureas and warfarin).54,154 The status of kidney function, particularly in elderly patients, is an important consideration before initiating treatment with metformin for a patient with AF.
Another biguanide, phenformin was withdrawn from use in most countries in 1976 due to the risk of lactic acidosis leading to severe metabolic acidosis.155 Although initially also considered a risk for metformin it is now clear that the incidence of lactic acidosis that can be directly linked metformin use is very low and usually only observed in patients with pre-existing co-morbidities such as significant renal and liver impairment, congestive heart failure, where its use should be monitored (see Metformin and Heart Failure), or resulting from metformin overdose.45,54,156
It is important to stress that lactic acidosis is also seen in people without diabetes and is increased in the presence of both hepatic and renal impairment, as well as serious infection, surgery, poor circulation, hypoxia, or excessive alcohol consumption, and therefore metformin should be discontinued and hemodialysis initiated if patients have a previous history or are symptomatic for lactic acidosis.144,157 However, even after 2016, the FDA approved the use of metformin in patients with mild-moderate chronic kidney disease there has been no evidence presented that metformin-associated lactic acidosis rates have increased.158
Metformin and Putative Protective Effects in AF
Several drug interactions with metformin should also be considered when prescribed to patients taking other drugs. For example, drug-induced changes in the levels of CYP450 enzymes may result in elevated levels of a wide range of drugs that then can compete with the cation transporters used by metformin for its bidirectional movement across cell membranes.54,159–162 Data from numerous pre-clinical and clinical studies have demonstrated the cardio-micro- and macro-vascular protective actions of metformin in reducing cardiovascular morbidity and mortality in patients with T2D and coronary artery disease.13,37,38,69 However, comparable evidence for patients without diabetes is sparse.69
Metformin is inexpensive and can potentially reduce the number of drugs a patient may need to take, thereby reducing the cost burden and increasing overall therapeutic compliance. However, before promoting its use in AF two important questions need to be answered:
1. Does metformin have benefits beyond its role as an anti-hyperglycaemic drug to reduce the risk of AF and treat AF? Thus, are the putative benefits of metformin in the prevention and treatment of AF simply secondary to its therapeutic efficacy as an antihyperglycemic drug and thereby reducing the known link between hyperglycemia/diabetes and increased risk of developing CVD?163
2. Does treatment with metformin also reduce the risk of AF in patients without diabetes?
To address these two questions, we evaluated data from both clinical and pre-clinical studies that have investigated the actions of metformin in humans and animal or cell-based protocols to determine whether there is support for repurposing metformin for use in AF.
Methods
Literature Search
For the purpose of evaluating the evidence regarding the efficacy of metformin therapy for AF, a narrative review of the literature published on Embase was conducted, facilitated by a librarian and co-author (RM). For this review, both peer reviewed original studies and reviews, along with non-peer reviewed (preprint) papers from bioRxiv and medRxiv etc., regarding the effects of metformin on AF were critically evaluated. Publications were selected based on evidence that the manuscripts support their claims with statistical evidence. Their scientific merit was also determined on the basis of their study design. Publications in Embase were identified by using the search strategy described in Table 2.
 |
Table 2 Search Strategy Used on Embase. The Search Was Based on “Metformin and AF and Diabetes and (Clinical Trials or Reviews)”. Total Number of Studies Used Was = 196 After Removing Duplicates (n = 3) |
The data presented in Figure 1 was generated from the Scopus database and based on the following search:
(TITLE-ABS-KEY (metformin OR dimethylbiguanidine OR dimethylguanylguanidine OR glucophage)) AND (TITLE-ABS-KEY (“atrial fibrillation” OR afib OR a-fib OR “Supraventricular tachycardia” OR svt OR arrhythmi* OR palpitat*))
Results
As summarised in Table 3 there is clinical evidence that supports the benefit of prescribing metformin to reduce the risk and/or symptoms of AF. However, the protective effects have not been universally shown, are not confined to metformin and extend to some other anti-diabetes drugs, particularly GLP-1 RAs. A 1998 study reported that patients taking a combination of metformin with other drugs experienced a significant decrease in the incidence of AF compared to those on metformin monotherapy, or to those who did not take any drugs. However, the number of patients in the metformin monotherapy group was low.24 The findings reported by Chang et al25 also emphasized that the protective effect of metformin monotherapy in patients newly diagnosed with T2D only lasted for 2–3 years during a 13-year follow-up period. This finding may indicate that the efficacy of using metformin for AF may be more pronounced during the earlier stages of diabetes and the development of AF. However, a comparison of metformin with other anti-diabetes agents was not made. In addition, based on studies with isolated atrial myocytes (HL-1 cells, derived from AT-1 mouse atrial cardiomyocyte tumor lineage), metformin (1 mM) reduced tachycardia-induced Reactive Oxygen Species (ROS) inferring that oxidative stress is an early trigger for AF.25 However, the effects of concentrations of metformin lower than 1 mM were not investigated and evidence of a direct negative chronotropic effect when metformin is used therapeutically have, to the best of our knowledge, not been reported.
 |
Table 3 Summary of clinical studies evaluating the effects of metformin in patients with AF. |
Other studies have compared the effect of metformin on AF to other newer agents. For example, meta-analysis of 5 clinical research studies comparing anti-diabetes drugs for the treatment of AF and atrial flutter showed that the GLP-1 RAs, in comparison to metformin, significantly reduced AF events in patients (OR 0.17, 95% CI 0.04–0.61).26 Chen et al27 reported that the use of metformin was not associated with an increased risk of new-onset AF (NAF); however, dipeptidyl peptidase (DPP4) inhibitors, also known as gliptins, were correlated with a reduced new-onset AF risk.27 Ostropoletes et al,29 in a retrospective study (with a total study population of 645,785 patients) compared metformin monotherapy and as a combination with other anti-diabetes drugs including sulfonylureas, thiazolidinediones (TZD), DPP4 inhibitors or GLP-1 RA. The study found that monotherapy with metformin significantly reduced the risk of AF when compared to DPP4 inhibitors (HR 0.90, 95% CI 0.84–0.96), sulfonylureas (HR 0.84, 95% CI 0.81–0.88) and TZD (HR 0.86, 95% CI 0.81–0.93). However, no significant benefit was seen when metformin monotherapy was compared to GLP-1 RAs (HR 0.95, 95% CI 0.86–1.05). Comparing metformin in combination with each of these anti-diabetes drugs, the study found that the combination of metformin with sulfonylurea was associated with a significantly higher AF risk when compared to a combination of metformin with DPP4 inhibitors (HR 1.2, 95% CI 1.1–1.4). However, inter-comparisons of metformin combination with TZD, GLP-1 RA or DPP4 and metformin and sulfonylurea with metformin and TZD or GLP-1 RA were not significant in terms of affecting the risk of AF.29
In a cohort study, Kim et al30 evaluated metformin monotherapy and combination therapy with other anti-diabetes drugs against AF. Metformin monotherapy or combination therapies were seen to reduce the risk of AF development. However, among all categories, metformin combination with TZD emerged as the most protective and effective as treatment for AF.30 Iqbal et al31 compared the use of monotherapy with metformin as first-line to monotherapy with another anti-diabetes drug as first-line in patients with T2D and found that metformin monotherapy did not significantly reduce the risk of developing AF.31 Zhou et al32 reported that when compared to monotherapy with metformin, monotherapy with sulfonylurea was associated with an increased risk of incident AF (HR 2.89, 95% CI 2.75–3.77).32 Treatment with metformin significantly reduced the risk of a recurrent atrial arrhythmia in patients with diabetes and AF after undergoing a catheter ablation procedure for their AF.164
Collectively, the data from observational clinical studies provide supportive, although not conclusive evidence, that metformin protects against AF. However, comparable benefits are also seen with some other anti-diabetes drugs, including GLP-1 RAs in humans.26,165 In addition, pre-clinical data with the db/db mouse model of T2D have shown that the GLP-1 RA, liraglutide, protects against AF.166 Thus, the question remains whether the benefit from metformin therapy is mediated as a result of its anti-hyperglycemic effects and low risk of hypoglycemia, or due to effects that offset the negative effects of the dysregulation of cardiac metabolism seen in diabetes, and/or the electrical, mechanical or anatomic remodeling mechanisms of AF itself.116,167 To answer this question, prospective data from placebo, double-blinded, randomized controlled clinical trials (RCTs) are needed. In this regard, the results of the REWIND trial with the GLP-1 RA, dulaglutide, failed to show the benefit of once weekly treatment on atrial arrhythmias in patient with diabetes.168
Mechanistic and Cellular Basis for the Benefits of Metformin in AF
AF is characterized by an irregularly irregular ventricular rhythm on the ECG, with atrial beats that can exceed 300 bpm, while ventricular heart rates remain at less than 200 bpm.41 The pathophysiology, cellular and molecular basis of AF has been extensively reviewed.169,170 AF is thought to develop as a result of atrial, electrical and electromechanical coupling and atrial remodeling, tissue fibrosis, and changes in ionic homeostasis, particularly with calcium handling in cardiac tissue as well as gap-junction proteins.169–171 Pre-existing hypertension is seen in 60–80% and diabetes in about 20% of patients with AF.172 In addition to age, other risk factors correlate with the development of AF in patients with diabetes include female sex, Caucasian race, obesity, hyperuricemia, non-alcoholic fatty liver disease, heart rate recovery and heart failure.173 The current therapy for AF includes rhythm control with antiarrhythmic drugs, catheter ablation, cryoballoon ablation, left atrial appendage closure, the Maze surgical procedure and anticoagulation therapy. In patients with a CHADS2VaSc score, which is linked to age, sex and pre-existing heart failure, hypertension, diabetes, stroke and vascular disease, equal to or more than 2 raises the risk for thromboembolic events such as stroke.174,175
Metformin, Glycemic Control and AF
The exact mechanism of how diabetes increases the risk of AF remains uncertain.176 Downstream sequalae that result from poor glycemic control include the dysregulation of cardiac metabolism, as well as electrochemical and structural remodelling that are known to enhance the risk of AF in patients with diabetes.116,177 Patients with diabetes are vulnerable to both episodes of hypoglycemia and hyperglycemia, especially if they are undergoing anti-diabetes treatment with agents such as sulfonylureas and insulin.178 Hypoglycemia in both T1D and T2D can also reduce the blood flow to the heart and cause disturbances in conduction, thereby increasing the risk of cardiovascular complications such as arrhythmias and prolonged QT interval.178 A prolonged QT interval, in addition to severe hypertension and hypokalemia that can result from hypoglycemia, is also associated with CVD, fatal arrhythmias and high mortality.179 Hypoglycemia also causes sympathetic activation, which is regarded as proarrhythmic.22 AF has been described as a complication of hypoglycemia in patients with diabetes.172 Hypoglycemia has also been shown to increase the susceptibility to AF in canine models and is associated with significantly more AF events than those seen with hyperglycemia.172 Thus, an obvious benefit of metformin is that its use, particularly as monotherapy, is associated with a 4.5-fold decrease in the risk of severe hypoglycemia in patients with diabetes, when compared to sulfonylureas, whereas the use of sulfonylureas is associated with a higher risk of AF.32,180
Diabetic cardiomyopathy is a consequence of uncontrolled hyperglycemia which results in apoptosis of cardiac myocytes and necrosis of the cardiac myocytes while stimulating the release of catecholamines from adrenocortical cells, resulting in an increased sympathetic activity.176 Severe episodes of hyperglycemia with sympathetic activation and reduced refractory period are associated with atrial remodelling and an increase in the risk of AF in these patients with diabetes.22 Excessive catecholamine release is also associated with increased insulin resistance, leading to impaired cardiac metabolism and function, and has a poor prognosis in patients with CVD, enhancing the risk of AF by triggering endoplasmic reticulum stress and myocardial fibrosis (see also Metformin AMPK and Cardiac Metabolism. On metformin and cardiac metabolism).181–183 Overall, good glycemic control reduces the incidence of AF and thus supports the recommendation to prescribe metformin to patients with diabetes who also are at risk of AF.17 Metformin use is regarded to be safe in AF and is a drug of choice in diabetic patients with cardiac arrhythmias.176 Although metformin therapy in patients with diabetes has been shown to be beneficial in the primary prevention of AF, intense glycemic control does not seem to confer the same advantages when compared to standard diabetes therapy.22,184 Furthermore, it is evident that, in general, anti-hyperglycemic agents that optimise glucose control should also benefit patients with AF.
Interestingly, glycemic fluctuations/excursions in the patient may contribute to AF-induced arrhythmogenesis, rather than just hypoglycemia or hyperglycemia alone. Supporting this possibility, glycemic fluctuations are positively associated with inflammation and atrial fibrosis, along with the upregulation of thioredoxin-interacting protein (Txnip), increasing the vulnerability of the heart to develop AF and new-onset AF.22 Glycemic fluctuations increase levels of oxidative stress as a result of increased ROS production due to increased NADPH oxidase activity and upregulated expression of Txnip, thus leading to cardiac fibrosis.172 Further, HbA1c variability over an extended period of time has also been correlated to an increased risk of AF development.185 Overall, the increased exposure and the severity of diabetes may dictate the onset and induction of AF in these patients.172 Consistent with this possibility, a case–control study in 2010 demonstrated that for every year a patient has diabetes, the risk of AF increases by 3%.17
Hyperglycemia and various other conditions, such as inflammation and aging, are also associated with the production and accumulation of advanced glycation end products (AGEs) that are linked to the development and progression of vascular disease in diabetes.186 Significantly high plasma AGE levels are found in patients with diabetes, and also in patients without diabetes. AGEs cross-link extracellular matrix proteins including type I collagen (expressed in atrial fibroblasts of patients with diabetes) and elastin and have been shown to lead to atrial fibrosis and senescence in diabetic mice.186,187 AGEs also stimulate inflammatory and oxidative responses by binding to their transmembrane receptors for AGEs (RAGE). Collectively, these factors contribute to AF-induced arrhythmogenesis in human subjects.188–190 Metformin has also been shown to chemically interact with methylglyoxal, a reactive dicarbonyl compound related to AGEs and levels of which are increased in diabetes, suggesting that metformin may also have direct effects to protect proteins against glycation by AGEs; however, whether this chemical interaction occurs when metformin is used clinically is unknown.191
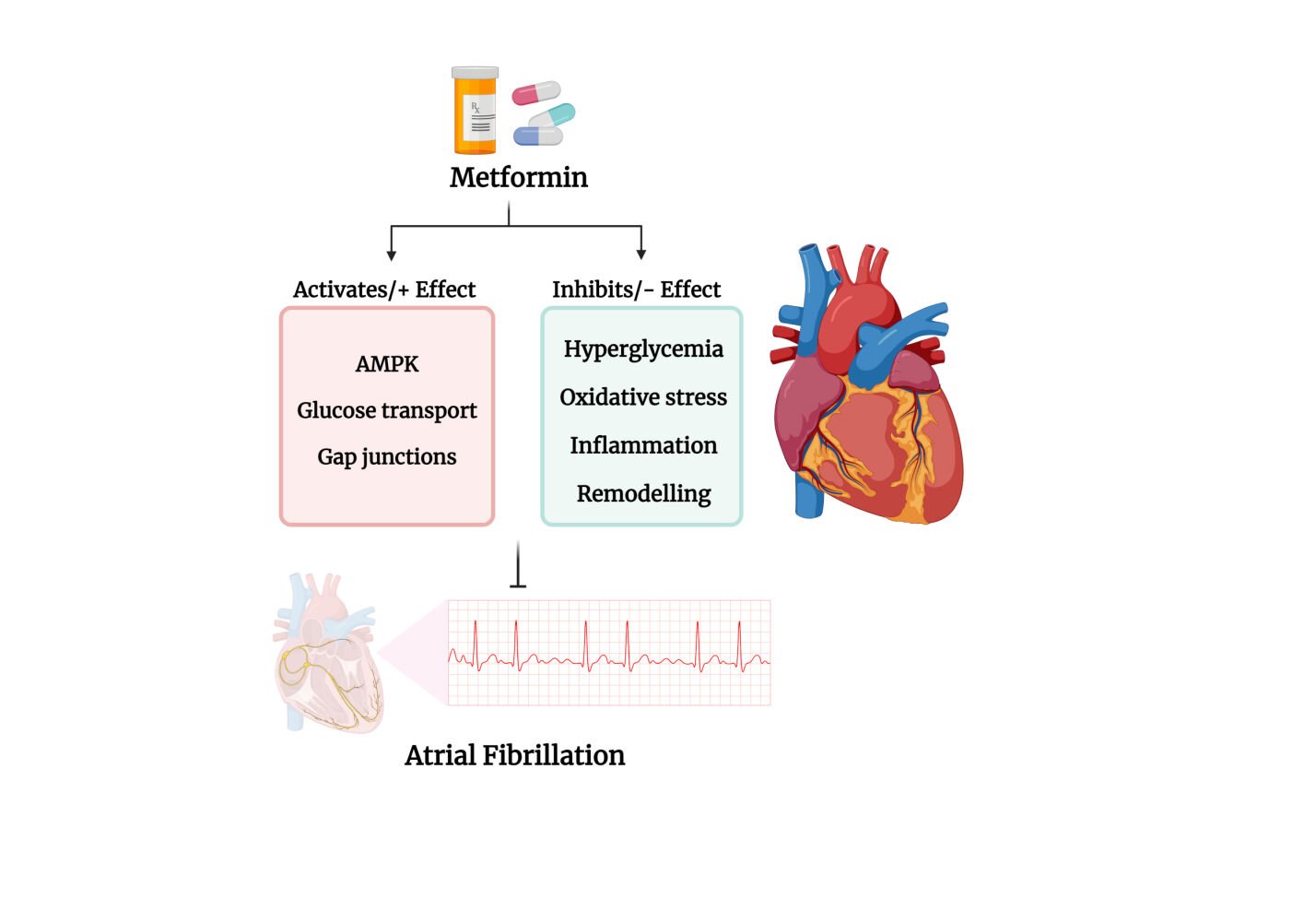
In summary, metformin by virtue of its proven positive effects to control hyperglycemia and insulin resistance result in downstream benefits to reduce oxidative stress and inflammation and can be predicted to have positive downstream effects on cardiac function and reduce the risk of AF.
Metformin and Heart Failure
Heart failure (HF) often accompanies AF and metformin reduces the risk of development of HF, atherosclerosis and coronary artery disease in patients and is considered a safe choice in patients with HF unless the HF is acutely decompensated, or if the patient has a GFR < 30 mL/min/1.73 m2.114,176 As stated earlier, the benefits of metformin may be due to the low risk of hypoglycaemia and improved glycemic control, as well as other effects such as improved vascular function via protective actions on the endothelium that has been demonstrated in numerous clinical and pre-clinical studies.13,38,175,192,193 However, other anti-diabetes drugs, alone or together with metformin, have also been shown to be effective – notably SGLT-2 inhibitors and GLP-1 RAs.176,179 In their 2020 review, Salvatore et al discuss the benefits of metformin as an adjunct to reduce the risk and treat HF, but also stress the need for prospective studies.58 Similarly, although the use of metformin to treat T2D with (at a dose of 1.5 g daily) is associated with a significant reduction in all-cause mortality, mortality due to cardiovascular events, non-fatal MI, non-fatal stroke, and arterial revascularization results in the context of treatment with the sulfonylurea, glipizide (30 mg daily), were not significantly different.194 These data are consistent with the benefits being associated with the anti-diabetes glucose-lowering actions of the agents rather than specifically to the class of drug.194 Similarly, treatment with metformin along with the thiazolidinedione (also known as glitazone or TZD), pioglitazone, or sulfonylurea, has been reported to be associated with a reduction in stroke, non-fatal MI and all-cause mortality.195
Possibly independent of metformin’s anti-hyperglycemic actions it has been hypothesized that metformin increases intracellular levels of adenosine which in turn prevents myocardial reperfusion injury by activating the adenosine receptor. The administration of 50 μM metformin in rats protected against myocardial reperfusion injury via the activation of adenosine receptors and protection was blocked by the adenosine antagonist, 8-(p-sulfophenyl)theophylline (8-SPT); however, the mechanism of action remains to be elucidated but does not involve effects on adenosine deaminase196 Others have also argued that adenosine may in part be responsible for the cardioprotective benefits of metformin.197 In atrial tissue, adenosine activates its A1 receptor in the sinoatrial node resulting in the Gβγ subunit-mediated opening of inward rectifying potassium channels (GIRK). When administered intravenously adenosine acts as a rapid onset and short-acting anti-arrhythmic drug to convert supraventricular arrhythmias to sinus rhythm.197–199
In summary, the use of metformin in patients with diabetes via its cardioprotective actions reduces the risk of HF that may accompany AF, however, data from prospective studies are required.
Metformin, AMPK and the Endothelium
The endothelium via pharmaco- and electromechanical coupling to the underlying vascular smooth muscle layer plays an essential role in the overall regulation of vascular tone, blood flow and conducted vasodilation in the microvasculature. In addition, the vascular endothelium generates an array of vasodilator agents, notably NO and prostacyclin, as well as vasoconstrictor molecules such as endothelin 2 and thromboxane A2. Disruption in the balance of locally acting vasodilator agents, that usually oppose vasoconstrictor molecules, is an important contributor to CVD.175,200–202 Metformin also reduces the levels of the endogenous inhibitor of eNOS, asymmetric dimethylarginine (ADMA). Elevated levels of ADMA are associated with diabetes and endothelial dysfunction and, in a rat model of hypertension, also elevated blood pressure.203,204
An accumulation of evidence indicates that the activation of AMPK by metformin is an important contributor to the cardiovascular protective benefits of metformin. Metformin activation of AMPK increases eNOS activity and NO-mediated cellular events that extend beyond direct effects on blood flow and including a positive effect on angiogenesis and revascularization. These combined actions have been argued to explain the protective effects of metformin in CVD, including HF, and may also extend to benefits for reducing the risk of AF.13,58,92,94,114,180,193,205–207
The anti-inflammatory effects of metformin may in part result secondarily to the activation of AMPK, eNOS and the enhanced generation of NO.148,208 AMPK-dependent effects include the reduction of lipotoxicity and enhancing autophagic flux.209,210 In addition, AMPK levels have been reported to be lower in cardiac tissue from animal models of AF, and the restoration of AMPK levels by metformin has been shown to reduce the infiltration of lipids and myofibril degradation in the diabetic heart.25,189,211 Furthermore, treatment with metformin is associated with a lower risk of MI when compared to insulin therapy.212 Activation of the AMPK-eNOS-NO pathway also activates peroxisome proliferator-activated receptor-gamma coactivator 1alpha (PGC-1α) that plays an important role in the regulation of energy metabolism, cardiovascular function and the biogenesis of mitochondria.213–215
It is important to note that in general, patients with AF are older with the addition of comorbidities such as diabetes, hypercholesterolemia, hypertension and other disorders, which also negatively affect endothelial function.175 AF is associated with endothelial dysfunction, but the mechanism is still unclear. However, the disruption of normal shear stress, laminar flow, and the resultant turbulent flow that results from the dysrhythmia are likely contributors. Such disturbed blood flow patterns are proposed to lead to eNOS down-regulation with reduced generation of NO resulting in hypercoagulability, as has been reported in patients with AF.216 NO also has strong antithrombotic effects in the arteries when released by activated platelets it prevents platelet recruitment, impedes plasminogen activator inhibitor-1 (PAI-1) activity and thus, prevents thrombus formation and development.217,218 Data from animal models of AF show a reduced expression of eNOS and low bioavailability of NO that arguably results from the low blood flow in the left atrium due to the dysregulation of atrial mechanical coupling.219 Data from patients also indicate that treatment with metformin lowers the levels of PAI-1 and stimulates the activity of the tissue plasminogen activator (TPA), decreasing the hypercoagulability state and increasing fibrinolysis, thereby attenuating a driver for AF.220 Furthermore, these effects appear to be independent of the impact of metformin on glycemic control.220 However, not all data from patients with and without AF support a role for the downregulation of eNOS as a major contributor; instead the data link the generation of ROS and its interaction with NO to produce cytotoxic peroxynitrite as key contributors to atrial myocyte dysfunction.221 A further link between AF and endothelial dysfunction is the increase in levels of von Willebrand factor (vWF), a large glycoprotein that is required for platelet adhesion, aggregation, and stabilization of Factor VIII (FVIII).222–224
In summary, collectively, these data support the argument that treatment with anti-diabetes drugs including metformin can have a benefit in reducing the risk and mortality associated with CVD such as AF but do not preclude that there are specific additional benefits that are linked to metformin’s cellular actions to protect endothelial-vascular function. In this context, metformin use may lower the risk of a stroke,84 although not all studies support this conclusion.225
Metformin, AMPK and Connexins
Intercellular communication via gap junction channels plays a critical role in the regulation of numerous cell processes including the coordinated contraction of cardiac tissue via facilitating the movement of small molecules including calcium and cyclic nucleotides, but not macromolecules.226 Gap junction channels are formed from hexamers of connexin proteins that come together to form connexons, which in the heart play an important role in the propagation of electrical signaling.227 There are three main connexins that are expressed in cardiomyocytes, among which, connexin 43 (Cx43) is expressed at the highest levels in the two ventricles. Cx40 and Cx43 are expressed at equal levels in the two atria, and Cx45 is expressed in the sinoatrial and atrioventricular node myocytes.228 Cx43 interacts with Zona Occludens-1 (ZO-1) to maintain the optimal physiological function of the gap junctions found between the adjacent cardiomyocytes thereby maintaining normal cardiac rhythm. An abnormal distribution, expression, and regulation by phosphorylation of connexins contributes to the structural and electrical remodelling of the atria and conduction abnormalities, increasing susceptibility to AF.172 Abnormalities in the expression of Cx43 proteins and remodelling of the gap junctions have been linked to cardiac arrhythmias and to slower conduction and re-entry pathways that trigger AF.229 In patients with diabetes, expression levels of Cx43 in the atrium are upregulated. However, phosphorylation of Cx43 is reduced significantly and thereby affecting both the gating of the connexin channels and the turnover of the gap junctions.172,230 Similarly, patients with diabetes are vulnerable to myocardial ischemia and diabetic cardiomyopathy and the expression of Cx43 is abnormally increased.231
Based on studies in transgenic mice, it has been found that the differential phosphorylation of Cx43 is important for the modulation of gap junction function at the intercalated disks and that remodelling of gap junctions contributes to arrhythmias by slowing the propagation of impulses. This remodelling may provide the basis for arrhythmogenesis.232 Mutations at the CK1 phosphorylation site of Cx43, S325/328/330, have been shown to be deleterious for gap junction assembly, size, turnover and optimal physiological function of the gap junctions. Such gap junction dysregulation can lead to electrical remodelling and a vulnerability to developing arrhythmias.230,233
The importance of AMPK and the regulation of connexin expression are supported by data from cardiac-specific LKB1-knockout mice wherein by the age of 12 weeks 97% have demonstrated spontaneous AF and show structural changes including bilateral atrial enlargement. Levels of ROS and inflammatory markers including CRP, interleukin 6, and tumor necrosis factor α are elevated, but there is decreased AMPK activity and reduced expression of Cx40 and Cx43 proteins.234 Data from cardiomyocytes from LKB1-knockout mice as well as cardiomyocytes from explanted hearts from patients with HF and AF have demonstrated evidence of mitochondria dysfunction as evidenced by reduced levels of ATP and changes in levels of markers of mitochondria function including mitochondrial electron transport chain, matrix, and inner and outer membrane proteins.235 Ozcan et al further demonstrated that treatment of cardiac specific LKB1-knockout mice with metformin (10 mg/kg/day) activated AMPK, and significantly reduced the incidence of AF; as well as the expression of connexin proteins.236 Interestingly, a report based on studies with diabetic rats has linked elevated AGEs to reduced phosphorylation of AMPK and dysregulation of Cx43 thereby promoting AF.170 Yang et al170 also reported that the AMPK agonist, 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR), reversed the effects of AGEs to down-regulate Cx43 expression.
Phosphorylation of Cx43 by protein kinase C-gamma (PKC-γ) regulates the assembly and disassembly of Cx43 and its interaction with ZO-1.237 PKC-γ-mediated serine phosphorylation of Cx43 causes disassembly of cardiac gap junctions, ie, loss of the gap junction Cx43 from the cell membrane or internalization of the gap junction Cx43.237 The interaction of Cx43 with ZO-1 is very important for converting the phosphorylation signals into structural and functional actions, while also maintaining the ability of Cx43 to respond to the activation of PKC-γ. Disruption of this interaction reduces the communication through these gap junctions as there is an increased retention of Cx43 on the interior of the cell membrane. Together, they are necessary to maintain the size, location, and structural integrity of these gap junctions and thus regulate the transduction pathway via phosphorylation by PKC-γ.238 Activation of AMPK by metformin, albeit based on the use of a high concentration of 200 μM, has been shown to modulate PKC-γ activity in human breast cancer cell lines239 and potentially may have a comparable effect in atrial tissue thereby potentially contributing to a protective action against AF.
The voltage-gated sodium channel Nav1.5 also interacts with Cx43 (but not ZO-1) in the perinexus of the cardiomyocytes and is important for impulse conduction between cells.240 LKB1 knockout mice also have a reduced expression of the cardiac Nav1.5 channel and show atrial remodeling that results in hypertrophic cardiomyopathy, thus reflecting an important role for LKB1 in both atrial structure, the maintenance of ionic homeostasis, and electro-mechanical coupling.241,242 Along with the reduction in Cx43 and Nav1.5 that was associated with the knockdown of LKB1, the transforming growth factor-beta (TGF-β) pathway was activated and an increased secretion of profibrotic factors were also seen, leading to fibrosis and increased susceptibility to AF.235,241,242
As previously discussed (Section 1.1.2), metformin has been reported to activate AMPK via the serine-threonine kinase LKB1.85 However, as an upstream regulator of AMPK, numerous cellular processes are also affected by LKB1, including glucose utilisation so the cardiac-specific knockdown of LKB1 would be expected to affect numerous cellular pathways. LKB1 also targets cellular pathways other than those regulated by AMPK and therefore some of the effects of metformin may be independent of AMPK activation including, for instance, an increase in the production of catalase and superoxide dismutase and suppression of Bax-dependent caspase 3 mediated apoptosis.242,243
In summary, collectively, data based on pre-clinical in vitro studies indicate that via the activation of AMPK, metformin may, at least in part, offset pathophysiological changes in the regulation of cardiac connexins and thereby reduce the risk of AF. Whether, these same mechanisms contribute to the benefits of metformin in treating patients with AF remains speculative (see Figure 4).
 |
Figure 4 Putative sites of action of metformin that contribute to its protective action against AF. Notes: (A) (Endothelial function): Metformin, via activation of adenosine monophosphate-activated protein kinase (AMPK) increases the bioactivity of endothelial nitric oxide synthase (eNOS) thereby increasing the generation of nitric oxide (NO). Metformin also reduces hypercoagulability and facilitates fibrinolysis by decreasing plasminogen activator inhibitor-1 (PAI-1) and increasing Tissue Plasminogen Activator (TPA). Additionally, metformin reduces endothelial dysfunction by inhibiting the production of Reactive Oxygen Species (ROS) and Advanced Glycation End-Products (AGE), as well as endothelial lipotoxicity. Lastly, metformin, in part via its vascular protective effects, reduces the risk of stroke. (B) (Connexins): Metformin activation AMPK which results not only in beneficial effects on glucose regulation and insulin resistance but also results in increase in the expression of Zonula Occludens-1 (ZO-1) and its interaction with and expression of connexin 43 (Cx43). (C) (Cardiac Dysmetabolism): Metformin, via its activation of AMPK, increases fatty acid and glucose uptake and fatty acid β-oxidation, while improving calcium homeostasis. Metformin also activates Peroxisome Proliferator-Activated Receptor α (PPARα) either directly or through AMPK, to increase fatty acid β-oxidation by increasing the activity of Very Long-Chain Acyl-Coenzyme A Dehydrogenase (VLCAD). In addition, metformin reduces triglyceride and fatty acid levels in the body and inhibits the accumulation of lipids in the left atrial (LA) appendage and the infiltration of lipids in the diabetic heart. (D) (Ion Channels): Metformin activation of AMPK results in the inhibition of the opening of ATP-sensitive potassium (KATP) channels, and it also increases the expression of voltage-gated sodium channels (Nav1.5) by activating serine/threonine kinase 11 (STK11/LKB11) (also known as liver kinase B1 (LKB1)), either through AMPK activation or its direct effect on Nav1.5 expression.35 (E) (Adenosine): Metformin activation of AMPK leads to an increase in intracellular adenosine which has cardioprotective effects via the adenosine A1 receptor. (F) (Oxidative Stress, Inflammation and Apoptosis): Metformin activation of AMPK via the STK11/LKB1 pathway results in the activation of eNOS, and increased production of catalase and superoxide dismutase which reduces ROS, and inhibits apoptosis. Metformin also decreases the generation of pro-inflammatory cytokines and inflammatory including Tumor Necrosis Factor-α (TNF- α), Interleukin-1β (IL-1β), Interleukin-6 (IL-6), C-Reactive Protein (CRP), Monocyte Chemoattractant Protein-1 (MCP-1), and Nuclear Factor-κB (NF-κB) and induces the polarization of macrophages towards anti-inflammatory M2 subtype over the pro-inflammatory M1 subtype).35,105,175 Created with BioRender.com. |
Metformin, AMPK, Mitochondria, ROS and Anti-Inflammatory Effects
The contribution of mitochondrial dysfunction to the pathophysiology of AF has been reviewed by Pool et al.244 Chronic treatment of mice with a high dose of metformin (300 mg/kg/day) reduces cardiac injury during ischemia-reperfusion and the effect, linked to the activation of AMPK, improved mitochondrial function and caused a reduction in endoplasmic reticulum (ER) stress.245 Support for the link between metformin and a role in the treatment of HF and AF via the protection of mitochondrial metabolism has been provided by independent studies in a mouse model of HF (coronary artery ligation) as well as clinical data from a randomized double-blinded study of metformin versus placebo in non-diabetic patients with HF.246,247 These data together with that summarized in reviews and systematic and retrospective meta-analysis of clinical cases, support the hypothesis that metformin has cardioprotective actions in patients with HF and coronary artery disease (CAD) that extend beyond those reported in patients with diabetes and result from improved cardiac metabolism and reduced oxidative stress.58,248 For instance, several studies have reported that metformin reduces mitochondrial ROS production and thereby ameliorates oxidative damage and apoptosis in the (atrial cell lysis and myolysis) heart, while increasing mitochondrial oxidative metabolism and Ca2+ levels.116,166 Similarly, metformin has been shown to reduce tachycardia-induced oxidative stress and myolysis in the heart in in-vitro experiments with mouse atrial myocytes 22 as well as reduce the ROS production due to tachypacing.25,189
Metformin, via the activation of AMPK and downstream effects, enhances SIRT3-mediated mitochondria biogenesis and protects against senescence in human aortic endothelial cells in culture.249 Evidence that metformin may have the same effects in patients with T2D is provided by data showing that treating patients with metformin at a dose of 1700 mg/day for at least one year corrected the expression of electron-chain complex proteins and reduces plasma levels of ROS in peripheral blood mononuclear cells (PBMCs) in 141 patients with T2D versus data from 101 healthy controls.250 Thus, in PBMCs from patients with T2D, levels of the mitochondrial serine/threonine PTEN-induced kinase 1 (PINK1) as well as the mitochondria quality control protein, Parkin, were reduced in comparison to those from controls; levels were restored after metformin treatment.251 Similarly, metformin restores PGC1α expression, indicating a role for metformin to enhance mitochondrial biogenesis and to protect cells from diabetes and stress-induced mitochondrial dysfunction.250 Comparable data for positive effects of metformin on PGC1α expression and mitochondrial biogenesis have been reported in rat and human hepatocytes treated in cell culture protocols with high (0.5 to 2 mM) concentrations of metformin.251
Pro-inflammatory biomarkers such as IL-6, IL-17, TNF-α and CRP that are elevated in patients with diabetes are associated with dilatation and structural remodelling of the atria and an increased incidence of AF.22 Pro-inflammatory mediators potentially can directly induce arrhythmias.252 These data suggest that the protective effects of metformin treatment against AF include the reduction of inflammatory factors such as CRP and IL-6.22,253 Furthermore, the pathways of glycolysis and fat oxidation, among others, are also associated with activation of the NLR family pyrin domain containing 3 (NLRP3) inflammasome in macrophages.254 A rise in the level of Ca2+ in the mitochondria also leads to NLRP3 inflammasome activation via increased ROS production. The inflammasome is a crucial inflammatory signaling complex, and its activation sensitizes the cardiac myocytes to inflammatory mediators, and causing them to become susceptible to spontaneous Ca2+ leaks and arrhythmogenic after depolarizations. Targeting the NLRP3 inflammasome signaling to treat AF is supported by results of a study of atrial cardiomyocytes from patients with AF wherein there was increased NLRP3 activity in AF.255 That metformin, via AMPK/mTOR signaling, can reduce NLRP3 activity is supported by cell culture data showing the effects of 2 mM metformin in cardiomyocytes from streptozotocin-induced diabetic mice treated with oral metformin (200 mg/day, for 8 days).256 The potential for targeting the NLRP3 inflammasome with metformin for the treatment of CVD has been reviewed in 2020 by Salvatore et al,58 and will not be discussed further in this review.
In summary, data from both pre-clinical and clinical studies indicate that metformin is associated with improvement in mitochondria function that can be argued to have beneficial effects in reducing inflammation and protect endothelial-vascular and cardiac function thereby reducing the risk of developing AF and HF.
Atrial Remodelling
Patients with diabetes are susceptible to structural and electrical remodelling of the atrium, which in turn enhances the risk of AF. Evidence from both cellular and clinical studies indicates that metformin can reduce the impact of remodeling and thereby reduce the risk of AF.35,172,257 Glycemic fluctuations, oxidative stress and inflammation go together in diabetes and contribute to the remodelling of the heart.22 Based on both animal and human studies, hyperglycemia has been linked to triggering remodelling.183,258,259 Additionally, activation of AGEs and RAGE and the RhoA and Rho-associated protein kinase (ROCK) pathway along with oxidative stress, inflammation and ROS production, and changes in connexin expression also contribute to the proarrhythmic remodelling of the atria.172,260 Increased pro-inflammatory factors such as TGF-β and connective tissue growth factor (CTGF) lead to increased atrial fibrosis, which in turn causes stiffening of the atria and the ventricles to contraction/relaxation, leading to diastolic dysfunction, and remodelling of the atria.22,172
Studies have also found that a large left atrial size, left ventricular hypertrophy and poor functioning of the left ventricle correlate with a high AF prevalence.189 Furthermore, studies show an association between diabetes and disordered arrangement and increased area of cross-section of the atrial cells, with the appearance of irregular myofibril arrangement, swollen, fragmented or vacuolated mitochondria and Z-lines that have degenerated.176 All of these factors contribute to the structural remodelling and increased susceptibility to AF development.
Diabetes is also associated with the proarrhythmic electrical and electrophysiologic remodelling of the atria. Diabetes is associated with the slowing of atrial conduction, along with the prolongation, spatial dispersion and changes in action potential duration (APD).261 Changes in the ionic currents for Na+, K+ and Ca2+ may also alter the conduction velocity or the morphology of the action potential in the atria, making the atria vulnerable to the development of AF.189 Data based on a study with diabetic rabbits showed a decrease in the Na+ current and an increase in L-type Ca2+ current linked to abnormalities in atrial conduction.176,183
In conclusion, metformin, by virtue of its anti-hyperglycemic actions, may help offset both structural and electromechanical remodelling and thereby contribute to reducing the risk of AF. This conclusion is supported by data from a study with a high fat and high sucrose-fed rat model of T2D, which shows that metformin corrects the expression levels of calcium-activated potassium channels SK2 (KCa2.2) and SK3 (KCa2.3).262,263 Additionally, metformin has been reported to reduce the production of ROS and other oxidative components such as AGEs, which adds to the efficacy of metformin to protect against endothelial dysfunction.264 Nantsupawat et al35 have reviewed the pre-clinical and clinical data that provides support for metformin targeting a number of pathways that prevent the structural remodelling of the atria and potentially contribute to the claimed beneficial effects of metformin in reducing the risk of AF (see Figure 5).
 |
Figure 5 Metformin reverses pathological remodeling. Notes: Putative targets for metformin to reverse pathological cardiac remodeling. (A) (Structural Remodeling): Metformin activation of adenosine monophosphate-activated protein kinase (AMPK) results in improved energy balance, improved mitochondrial function, inhibition of fibrosis, and reduced generation of Reactive Oxygen Species (ROS). AMPK also corrects gap junction expression and improves cell-to-cell coupling. Metformin reduces the generation of pro-inflammatory mediators leading to reduction in fibrosis and degradation of myofibrils in the diabetic heart. Lastly, metformin also reduces the production of Advanced Glycation End-Products (AGE) and Receptors for Advanced Glycation End-Products (RAGE) by counteracting hyperglycemia, which also reduces ROS and tissue fibrosis. (B) (Electrical Remodeling): In animal models as well as in humans metformin reduces electrical remodeling by improving ionic homeostasis via the activation of AMPK. (C) (Electromechanical Remodeling): Metformin has direct effects to improve ionic homeostasis and inhibit fibrosis and reduce electromechanical remodeling. (D) (Autonomic Remodeling): Metformin, via its anti-diabetes actions, reduces the risk of autonomic neuropathy thereby reducing remodeling of the heart.116,166,168–170 Created with BioRender.com. |
Metformin, AMPK and Cardiac Metabolism
Cardiac muscle is active continuously under aerobic conditions (oxidative phosphorylation), and relies mainly on fatty acid oxidation (approximately 95%), over glucose oxidation (approximately 5%) as a source of energy, due to the relative efficiency of the former vs the latter in terms of ATP production.265,266 In brief, fatty acid uptake into cardiomyocytes occurs through plasma membrane protein transporters that involve fatty acid translocase (FAT)/CD36 and fatty acid-binding proteins (FABP).267 Fatty acids are oxidized in the mitochondria (β-oxidation) forming acetyl-CoA which is then used as a substrate in the Tricarboxylic Acid Cycle (TCA).268 Glucose uptake into cardiomyocytes occurs via GLUT1 and GLUT4 transporters and is oxidized via glycolysis to produce pyruvate as the end-product, along with ATP. Pyruvate is converted to acetyl-CoA by the pyruvate dehydrogenase complex (PDC), which is fed into the TCA cycle.269 The heart can also utilize, to a limited extent, ketone bodies, which in a normal heart may provide 10–15% of the ATP supply.270
In pathologic states, such as AF, cardiac metabolism is impaired and this predisposes the heart to atrial arrhythmias, which in turn negatively affect metabolic activity, thus contributing to a downward spiral that further enhances metabolic dysregulation. The role of the dysregulation of cardiac metabolism to arrhythmogenesis and the mechanisms and potential benefits of metformin have been reviewed by Lkhagva et al.116 Cardiac disease is also associated with a greater contribution of ketone bodies to energy supply.271,272 In addition, analysis of transcriptional data from patients with AF indicates there is a downregulation of the enzymes that regulate fatty acid oxidation and a compensatory upregulation of the enzymes involved in glucose utilization.273 In patients with new onset AF, as for instance with post-operative AF, there is a decrease in expression of the fatty acid-binding protein, FABP3, and this decrease is associated with a lower level of fatty acid uptake and oxidation.274 In contrast, chronic AF is associated with a higher expression of FABP3 and an increase in the uptake of fatty acids into myocytes.275 As a consequence, patients with chronic AF have higher levels of free fatty acids in the serum and a higher expression of genes involved in transport uptake of fatty acids into the atria. The increased uptake promotes arrhythmogenesis of AF in these patients.276 Furthermore, elevated plasma levels of fatty acids as well as glucose inhibit Na+/K+ ATPase, resulting in an increase in intracellular Na+ and Ca2+ that can induce cardiac dysfunction and arrhythmias.277–279
In patients with T2D, the risk of AF is enhanced as insulin resistance reduces glucose uptake and utilization, while increasing fat oxidation, thereby decreasing the flexibility to obtain energy from alternative substrates, and enhancing the susceptibility of the heart to ischemia and arrhythmogenesis. Supportive evidence is provided from studies with regular versus irregular paced rat cardiomyocytes as well data from mouse and human atrial myocytes-paced models of AF that indicate that the transport of glucose into cardiomyocytes is reduced due to a lower expression of GLUT4 transporters and a reduced expression of the protein SNAP-23, which is required in the translocation of GLUT4 to the plasma membrane.280 The reduced expression and translocation of glucose transporters is associated with an increase in fat uptake into these cells as a result of the increased expression of FAT/CD36 in the membrane, thus further enhancing the downward spiral of cardiac metabolism dysregulation.281 A correlation has also been shown between infiltration of adipose tissue in the epicardium of patients with diabetes, which can contribute to the development of AF.282
As a result of the accumulation of fatty acids inside cardiomyocytes, ROS levels and lipotoxicity are enhanced, and this increase in ROS results in mitochondrial dysfunction, which also occurs with aging.58,281 The overproduction of ROS results in the oxidation and activation of calmodulin-dependent protein kinase II (CaMKII) at methionines 281/282, which is involved in the regulation of Ca2+ handling, resulting in Ca2+ dysregulation and arrhythmogenesis.283 The levels of GLUT4, PPARα, sirtuin-1, mast cell protease 1 (mCPT-1), medium-chain acyl-CoA dehydrogenase, and peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1α) are substantially downregulated, signifying a reduced glucose uptake.284 In patients with short-term AF, such as paroxysmal AF (intermittent, spontaneous termination or within a week of treatment) and short-lasting persistent AF (does not terminate within a week), mitochondria are able to adapt to the ATP demand through oxidative phosphorylation.116,285 However, in patients with longstanding AF, the mitochondria fail to cope, leading to a diminished ATP level. The reduction in intracellular ATP levels affects enzymatic reactions and the ionic balance across membranes, impairing the ionic homeostasis and contractions of the myocytes.58 This reduction in ATP also leads to an increased compensation via anaerobic glycolysis, resulting in an increased lactate production, similar to the metabolic stress related to the Warburg effect. Studies have detected increased levels of lactate in the cytosol and inhibition of pyruvate dehydrogenase by kinases suggesting that pyruvate is increasingly converted into lactate rather than acetyl-CoA, in a context of anaerobic glycolysis/Warburg effect in the cardiomyocytes and the uncoupling of glycolysis from mitochondrial oxidation of pyruvate in AF.286 Increased lactate accumulation has been shown to promote arrhythmogenesis and thereby contribute to the development of AF through the dysregulation of the electrophysiological characteristics of the atrial cardiac cells.287–289
In canine models of AF, levels of very-long chain acyl-CoA dehydrogenase (VLCAD) the rate-limiting enzyme in the β-oxidation of fatty acids in the mitochondria are reduced. In this context, metformin can reduce fat accumulation by upregulating the AMPK/PPARα/VLCAD pathway.253 It remains unclear if this effect is AMPK-dependent or independent. However, AMPK activates the peroxisome proliferator-activated receptor α (PPARα), which in turn activates the VLCAD to stimulate β-oxidation of fatty acids, thereby providing another potential mechanism whereby metformin may reduce AF.253 Metformin reduces the accumulation of lipids and the levels of triglycerides and free fatty acids in the left atrial appendage and stimulates the uptake and β-oxidation of fatty acids, while reducing the synthesis of triglycerides and fatty acids, and inhibits both ACC 1 and 2 through the same AMPK/PPARα/VLCAD pathway.290
In conclusion, metformin, via the activation of AMPK, stimulates uptake of both glucose via GLUT4 transporters, and β-oxidation of fatty acids while reducing the synthesis of triglycerides and fatty acids, and inhibiting both ACC 1 and 2; this provides strong support for a protective action of metformin against AF.116,290,291 Treatment of patients with T2D with metformin has also been reported to increase adiponectin levels, which in turn increase insulin sensitivity and β-oxidation of fats and reduce hepatic glucose synthesis; this reduces metabolic dysfunction and the risk of AF.292 However, the relationship between adiponectin levels and AF may be U-shaped as high levels of adiponectin have been linked to a heightened risk of AF, requiring further investigation.293
In summary, treatment with metformin via its AMPK-mediated effects on cardiac metabolism can correct metabolic dysfunction and thereby reduce the risk of AF.
Limitations
Our study has Limitations. Although we reviewed an extensive literature base such is the interest in the potential for repurposing metformin for multiple diseases, including AF, it was not possible to provide equity and critically evaluate and cite all of the studies with the same level of emphasis. Another limitation, is that care is required in extrapolating data from pre-clinical studies with metformin to the clinical setting. To offset this limitation we have, where appropriate, provided a critique of the protocols used in these studies. Furthermore, as has been emphasised, there are very few studies that address the effects of metformin in patients without diabetes, thus potentially biasing our conclusions in favour of a pleiotropic effect of metformin in reducing the risk of AF. However, we have been careful to repeatedly address this issue by providing the details of the studies as well as the limitations of the studies that we have cited.
Conclusions
There is a general concordance based on extensive data primarily from pre-clinical studies that metformin, via the activation of both AMPK-dependent and AMPK-independent signaling pathways, has multiple actions on glucose and lipid metabolism as well as cardiac and endothelial-vascular function that could contribute to reducing the risk of diabetes-associated CVD, including the risk of AF. Figure 6 provides a summary of the major benefits of metformin that may offset AF and support the use of repurposing metformin to treat AF. Data from pre-clinical studies also provide evidence that metformin via a number of cellular mechanisms that include effects on cardiac gap junctions can reduce the risk of AF by reducing the risk of arrhythmogenesis. A number of observational studies (see Table 1 and25,27,28) also provide support for a protective effect of metformin against the development of AF thereby supporting the argument that treatment with metformin would be a useful adjunct in the management of AF. However, data from the same clinical studies show that the benefits were not sustained in older (>65 years old) patients. In addition, to date there is also a lack of sufficient supportive data from prospective RCT double-blinded studies. One such RCT study, which investigated the effects of metformin on the occurrence of arrhythmias post coronary artery bypass surgery, failed to show a benefit of metformin over placebo.294 Interestingly, the conclusions are similar for GLP-RAs where pre-clinical and data from observational clinical data are supportive of protection against the development of AF, whereas data from prospective studies, such as REWIND, have failed to show a benefit over placebo.167 The lack of positive data from RCTs needs to be tempered by the fact that to date relatively few studies have been completed and therefore a definitive conclusion cannot be reached at this time. Nevertheless, for the majority of patients with T2D, metformin is a safe drug that has proven benefits in terms of improving glycemic control with a minimal risk of hypoglycemia and has cardiovascular protective actions and is therefore appropriate to be used in patients with T2D who are also at risk of AF. At the same time (as is the case for all drugs) there are potential side effects associated with the chronic use of metformin such as vitamin B12 deficiency plus its associated neuropathy and elevated risk of AF and other cardiac arrhythmias.149,153 Concerns over use in pregnancy and effects on fetal growth have also been expressed,130–133 as well as the environmental effects of metformin when it ends up in wastewater, which, as we have discussed, have been linked to potential effects on human reproductive health.126 Finally, it remains uncertain whether metformin’s multiple cellular effects via AMPK extend to benefits in patients without diabetes, and additional data is needed from appropriately designed RCTs.
 |
Figure 6 Metformin as a treatment for AF. Notes: In this schematic metformin is depicted to have multiple beneficial effects that collectively lower the risk of AF and arguably justify its repurposing as an adjunct therapeutic drug for the treatment of atrial fibrillation (AF). A long history of clinical use indicates that metformin, via its antihyperglycemic and insulin sensitizing actions, is a safe drug for the treatment of type 2 diabetes that unlike sulfonylureas and insulin is not associated with the risk of hypoglycemia. These actions as an anti-diabetes agent may entirely explain metformin’s putative benefits in AF including the reduction of oxidative stress, cardiac metabolism dysregulation, inflammation and decreased fibrosis and structural remodeling; however, pleiotropic actions of metformin may also contribute. An accumulation of evidence supports the hypothesis that metformin’s cellular actions are mediated, at least in part, via the activation of 5’ adenosine monophosphate-activated protein kinase (AMPK), either directly or via activation of upstream kinases such as liver kinase B1 (LKB1). AMPK has multiple downstream targets that include: (i) Effects on cardiac connexins and ion channels that can improve electrical conduction in the heart. (ii) Enhanced activity of endothelial nitric oxide synthase (eNOS) and generation of nitric oxide (NO) that has multiple positive effects to improve endothelial function and reduce cardiac and vascular dysfunction that collectively can reduce the risk of AF. (iii) A reduction of oxidative stress in part via its antihyperglycemic actions and beneficial effects on fatty acid uptake and metabolism in the heart as well as also via direct effects on mitochondrial function. (iv) Collectively, these actions result in a reduced inflammation, and beneficial effects to offset atrial remodeling. Created with BioRender.com. |
Abbreviations
ACC-2, acetyl-CoA carboxylase; ADMA, Asymmetric dimethylarginine; AF, Atrial fibrillation; AGE, Advanced Glycation End-Product; AICAR, 5-aminoimidazole-4, carboxamide ribonucleoside; AMPK, AMP-activated protein kinase; ATM, Ataxia Telangiectasia Mutated gene; CaM kinase II, Calmodulin-dependent protein kinase II; CK1, Casein kinase 1; CTGF, Connective tissue growth factor; CRP, C-Reactive Protein; Cx, connexin; CVD, Cardiovascular disease; DPP-4, dipeptidyl peptidase 4 inhibitors; eNOS, endothelial nitric oxide synthase; ER, Endoplasmic reticulum; FABP, Fatty acid-binding protein; GLP-1 RAs, Glucagon-like peptide receptor agonists; GLUT1, Glucose transporter type 1; GLUT4, Glucose transporter type 4; GFR, glomerular filtration rate; IL-1β, Interleukin-1β; LKB1/STK11, liver kinase B1/serine threonine kinase 11; mTOR – mammalian target for rapamycin; MCP-1, Monocyte Chemoattractant Protein-1; NF-κB, Nuclear Factor-κB; NLRP3, NOD-like receptor (NLR) family pyrin domain containing 3 inflammasome; NO, nitric oxide; NR4A1 (Nur77), Orphan Nuclear Receptor Subfamily 4 Group A Member 1; OCT, organic cation transporters; PAI-1, plasminogen activator inhibitor-1; PBMCs, peripheral blood mononuclear cells; PGC-1α, Peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1alpha; PKC-γ, Protein Kinase C-γ; PPARα, Peroxisome Proliferator-Activated Receptor α; PI3/PI4, phosphatidylinositol 3/phosphatidylinositol 4; PCOS, polycystic ovary syndrome; RAGE, Receptor for Advanced Glycation End-Product; RCT, randomized controlled trial; ROCK, RhoA-associated protein kinase; ROS, Reactive Oxygen Species; SGLT-2, sodium-glucose co-transporter-2 inhibitors; Sirtuin −1 NAD-dependent deacetylase; T2D, type 2 diabetes; TCA, Tricarboxylic Acid Cycle; TGF-β, Transforming Growth Factor-β; TPA, Tissue Plasminogen Activator; TNFα, Tumor Necrosis Factor-α; TZD, thiazolidinediones, also known as glitazones; UKPDS, United Kingdom Prospective Diabetes Study; VLCAD, Very Long-Chain Acyl-Coenzyme A Dehydrogenase; ZO-1, Zonula Occludens-1.
Acknowledgments
Figures were created using BioRender.com license with the agreement number: DC26U1YG8B.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all of these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Isra Marei was supported by the L’Oreal-UNESCO For Women in Science Young Talents Program/Middle East, 2020; Kareem Fanous received support from a Medical Student Research Award (MSRA) from Weill Cornell Medical College in Qatar.
Disclosure
Mr Kareem Fanous reports grants from Qatar National Research Fund, during the conduct of the study. All authors declare no other conflicts of interest.
References
1. Roth GA, Mensah GA, Johnson CO, et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: update From the GBD 2019 Study. J Am Coll Cardiol. 2020;76(25):2982. doi:10.1016/j.jacc.2020.11.010
2. WHO Cardiovascular diseases (CVDs). Available from: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds).
3. Amini M, Zayeri F, Salehi M. Trend analysis of cardiovascular disease mortality, incidence, and mortality-to-incidence ratio: results from global burden of disease study 2017. BMC Public Health. 2021;21:401. doi:10.1186/s12889-021-10429-0
4. Garattini S, Bertele’ V. Efficacy, safety and cost of new cardiovascular drugs: a survey. Eur J Clin Pharmacol. 2008;59(8–9):701–706. doi:10.1007/s00228-003-0634-y
5. Braunwald E. Gliflozins in the Management of Cardiovascular Disease. N Engl J Med. 2022;386(21):2024–2034. doi:10.1056/NEJMra2115011
6. Kosiborod MN, Abildstrøm SZ, Borlaug BA, et al. STEP-HFpEF Trial Committees and Investigators. Semaglutide in Patients with Heart Failure with Preserved Ejection Fraction and Obesity. N Engl J Med. 2023;389(12):1069–1084. doi:10.1056/NEJMoa2306963
7. Lincoff AM, Brown-Frandsen K, Colhoun HM, et al. SELECT Trial Investigators. Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes. N Engl J Med. 2023;389(24):2221–2232. doi:10.1056/NEJMoa2307563
8. Ozawa S, Shankar R, Leopold C, et al. Access to medicines through health systems in low- and middle-income countries. Health Policy Plan. 2019;34(Supplement_3):iii1–iii3. doi:10.1093/heapol/czz119
9. Gaziano TA, Bitton A, Anand S, et al. Growing epidemic of coronary heart disease in low- and middle-income countries. Curr Probl Cardiol. 2010;35(2):72–115. doi:10.1016/j.cpcardiol.2009.10.002
10. Granger CB, Pocock SJ, Gersh BJ. The need for new clinical trials of old cardiovascular drugs. Nat Rev Cardiol. 2022;20(2):71–72. doi:10.1038/s41569-022-00819-1
11. Samuel SM, Varghese E, Kubatka P, et al. Metformin: the Answer to Cancer in a Flower? Current Knowledge and Future Prospects of Metformin as an Anti-Cancer Agent in Breast Cancer. Biomolecules. 2019;9(12):846. doi:10.3390/biom9120846
12. Mohammed I, Hollenberg MD, Ding H, Triggle CR. A Critical Review of the Evidence That Metformin Is a Putative Anti-Aging Drug That Enhances Healthspan and Extends Lifespan. Front Endocrinol (Lausanne). 2021;2:718942. doi:10.3389/fendo.2021.718942
13. Triggle CR, Mohammed I, Bshesh K, et al. Metformin: is it a drug for all reasons and diseases? Metabolism. 2022;133:155223. doi:10.1016/j.metabol.2022.155223
14. Pushpakom S, Iorio F, Eyers PA, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2019;18(1):41–58. doi:10.1038/nrd.2018.168
15. Dal Canto E, Ceriello A, Rydén L, et al. Diabetes as a cardiovascular risk factor: an overview of global trends of macro and micro vascular complications. European Journal of Preventive Cardiology. 2019;26(2_suppl):25–32. doi:10.1177/2047487319878371
16. Benjamin EJ, Levy D, Vaziri SM, et al. Independent risk factors for atrial fibrillation in a population-based cohort. The Framingham Heart Study. JAMA. 1994;271(11):840–844. doi:10.1001/jama.1994.03510350050036
17. Dublin S, Glazer NL, Smith NL, et al. Diabetes mellitus, glycemic control, and risk of atrial fibrillation. J Gen Intern Med]. 2010;25(8):853–858. doi:10.1007/s11606-010-1340-y
18. Huxley RR, Filion KB, Konety S, Alonso A. Meta-analysis of cohort and case-control studies of type 2 diabetes mellitus and risk of atrial fibrillation. Am J Cardiol. 2011;108(1):56–62. doi:10.1016/j.amjcard.2011.03.004
19. Perez MV, Wang PJ, Larson JC, et al. Risk factors for atrial fibrillation and their population burden in postmenopausal women: the Women’s Health Initiative Observational Study. Heart. 2013;99(16):1173–1178. doi:10.1136/heartjnl-2013-303798
20. Qi W, Zhang N, Korantzopoulos P, et al. Serum glycated hemoglobin level as a predictor of atrial fibrillation: a systematic review with meta-analysis and metaregression. PLoS One. 2017;12(3):e0170955. doi:10.1371/journal.pone.0170955
21. Sun Y, Hu D. The link between diabetes and atrial fibrillation: cause or correlation? J Cardiovasc Dis Res. 2010;1(1):10–11. doi:10.4103/0975-3583.59978
22. Wang A, Green JB, Halperin JL, et al. Atrial Fibrillation and Diabetes Mellitus: JACC Review Topic of the Week. J Am Coll Cardiol. 2019;74(8):1107–1115. doi:10.1016/j.jacc.2019.07.020
23. Xiong Z, Liu T, Tse G, et al. A machine learning aided systematic review and meta-analysis of the relative risk of atrial fibrillation in patients with diabetes mellitus. Front Physiol. 2018;9:835. doi:10.3389/fphys.2018.00835
24. Davis TME, Parsons RW, Broadhurst RJ, et al. Arrhythmias and Mortality After Myocardial Infarction in Diabetic Patients: relationship to diabetes treatment. Diabetes Care. 1998;21(4):637–640. doi:10.2337/diacare.21.4.637
25. Chang SH, Wu LS, Chiou MJ, et al. Association of metformin with lower atrial fibrillation risk among patients with type 2 diabetes mellitus: a population-based dynamic cohort and in vitro studies. Cardiovasc Diabetol. 2014;13(1):1–8. doi:10.1186/s12933-014-0123-x
26. Shi W, Zhang W, Zhang D, et al. Comparison of the effect of glucose-lowering agents on the risk of atrial fibrillation: a network meta-analysis. Heart Rhythm. 2021;18(7):1090–1096. doi:10.1016/j.hrthm.2021.03.007
27. Chen HY, Yang FY, Jong GP, Liou YS. Antihyperglycemic drugs use and new-onset atrial fibrillation in elderly patients. Eur J Clin Invest. 2017;47(5):388–393. doi:10.1111/eci.12754
28. Liou YS, Yang FY, Chen HY, Jong GP. Antihyperglycemic drugs use and new-onset atrial fibrillation: a population-based nested case control study. PLoS One. 2018;13(8):e0197245. doi:10.1371/journal.pone.0197245
29. Ostropolets A, Elias PA, Reyes MV, et al. Metformin Is Associated with a Lower Risk of Atrial Fibrillation and Ventricular Arrhythmias Compared to Sulfonylureas: an Observational Study. Circ Arrhythm Electrophysiol. 2021;14(3):e009115. doi:10.1161/CIRCEP.120.009115
30. Kim S, Park SY, Kim B, et al. Association between antidiabetic drugs and the incidence of atrial fibrillation in patients with type 2 diabetes: a nationwide cohort study in South Korea. Diabet Res Clin Pract. 2023:198. doi:10.1016/j.diabres.2023.110626
31. Iqbal A, Tekin Z, Kattan MW, et al. Association between first-line monotherapy with metformin and the risk of atrial fibrillation (AMRAF) in patients with type 2 diabetes. J Diabetes Complications. 2022;36(11):108315. doi:10.1016/j.jdiacomp.2022.108315
32. Zhou J, Zhang G, Chang C, et al. Metformin versus sulphonylureas for new onset atrial fibrillation and stroke in type 2 diabetes mellitus: a population-based study. Acta Diabetol. 2022;59(5):697–709. doi:10.1007/s00592-021-01841-4
33. Lal JC, Mao C, Zhou Y, et al. Transcriptomics-based network medicine approach identifies metformin as a repurposable drug for atrial fibrillation. Cell Reports Med. 2022;3(10):100749.
34. Vinciguerra M, Olier I, Ortega-Martorell S, et al. New use for an old drug: metformin and atrial fibrillation. Cell Reports Med. 2022;3(12):100875. doi:10.1016/j.xcrm.2022.100875
35. Nantsupawat T, Wongcharoen W, Chattipakorn SC, Chattipakorn N. Effects of metformin on atrial and ventricular arrhythmias: evidence from cell to patient. Cardiovasc Diabetol. 2020;19(1):198. doi:10.1186/s12933-020-01176-4
36. Li JZ, Li YR. Cardiovascular Protection by Metformin: latest Advances in Basic and Clinical Research. Cardiology. 2023;148(4):374–384. doi:10.1159/000531432
37. Bailey CJ. Metformin: effects on micro and macrovascular complications in type 2 diabetes. Cardiovasc Drugs Ther. 2008;22(3):215–224. doi:10.1007/s10557-008-6092-0
38. Triggle CR, Marei I, Ye K, et al. Repurposing Metformin for Vascular Disease. Curr Med Chem. 2023;30(35):3955–3978. doi:10.2174/0929867329666220729154615
39. Preiss D, Lloyd SM, Ford I, et al. Metformin for non-diabetic patients with coronary heart disease (the CAMERA study): a randomised controlled trial. Lancet Diabetes Endocrinol. 2014;2(2):116–124. doi:10.1016/S2213-8587(13)70152-9
40. Miyasaka Y, Barnes ME, Gersh BJ, et al. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation. 2006;114(2):119–125. doi:10.1161/CIRCULATIONAHA.105.595140
41. Nayak S, Natarajan B, Pai RG. Etiology, Pathology, and Classification of Atrial Fibrillation. Int J Angiol. 2020;29(2):65–71. doi:10.1055/s-0040-1705153
42. Bailey CJ. Metformin: historical overview. Diabetologia. 2017;60(9):1566–1576. doi:10.1007/s00125-017-4318-z
43. Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996;334(9):574–579. doi:10.1056/NEJM199602293340906
44. Witters LA. The blooming of the French lilac. J Clin Invest. 2001;108(8):1105–1107. doi:10.1172/JCI14178
45. Scarpello JH, Howlett HC. Metformin therapy and clinical uses. Diab Vasc Dis Res. 2008;5(3):157–167. doi:10.3132/dvdr.2008.027
46. Khilnani G. Metformin: a Reflection on My Journey as Antidiabetic Drug. GAIMS Journal of Medical Sciences. 2023;3(2):34–40.
47. Viollet B, Guigas B, Sanz Garcia N, et al. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond). 2012;122(6):253–270. doi:10.1042/CS20110386
48. Bosi E. Metformin—The Gold Standard in Type 2 Diabetes: what Does the Evidence Tell Us? Diabetes Obesity Metab. 2009;11,3–8. doi:10.1111/j.1463-1326.2008.01031.x
49. Foretz M, Hébrard S, Leclerc J, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120(7):2355–2369. doi:10.1172/JCI40671
50. Gu S, Tang Z, Shi L, et al. Cost-Minimization Analysis of Metformin and Acarbose in Treatment of Type 2 Diabetes. Value Heal Reg Issues. 2015;6:84–88. doi:10.1016/j.vhri.2015.03.012
51. Glossmann HH, Lutz OMD. Pharmacology of metformin - An update. Eur J Pharmacol. 2019;865:172782. doi:10.1016/j.ejphar.2019.172782
52. He L. Metformin and Systemic Metabolism. Trends Pharmacol Sci. 2020;41(11):868–881. doi:10.1016/j.tips.2020.09.001
53. LaMoia TE, Shulman GI. Cellular and Molecular Mechanisms of Metformin Action. Endocr Rev. 2022;42(1):77–96. doi:10.1210/endrev/bnaa023
54. Corcoran C, Jacobs TF. Metformin. 2023. Treasure Island (FL): StatPearls Publishing; 2024.
55. Johnson NP. Metformin use in women with polycystic ovary syndrome. Annals of Translational Med. 2014;2(6):56. doi:10.3978/j.issn.2305-5839.2014.04.15
56. Pryor R, Cabreiro F. Repurposing metformin: an old drug with new tricks in its binding pockets. Biochem J. 2015;471(3):307–322. doi:10.1042/BJ20150497
57. Samuel SM, Varghese E, Büsselberg D. Therapeutic Potential of Metformin in COVID-19: reasoning for Its Protective Role. Trends Microbiol. 2021;29(10):894–907. doi:10.1016/j.tim.2021.03.004
58. Salvatore T, Pafundi PC, Galiero R, et al. Can Metformin Exert as an Active Drug on Endothelial Dysfunction in Diabetic Subjects? Biomedicines. 2020a;9(1):3. doi:10.3390/biomedicines9010003
59. Salvatore T, Pafundi PC, Morgillo F, et al. Metformin: an old drug against old age and associated morbidities. Diabet Res Clin Pract. 2020b;160:108025. doi:10.1016/j.diabres.2020.108025
60. Varghese E, Samuel SM, Liskova A, et al. Diabetes and coronavirus (SARS-CoV-2): molecular mechanism of Metformin intervention and the scientific basis of drug repurposing. PLoS Pathog. 2021;17(6):e1009634. doi:10.1371/journal.ppat.1009634
61. Martin DE, Cadar AN, Bartley JM. Old drug, new tricks: the utility of metformin in infection and vaccination responses to influenza and SARS-CoV-2 in older adults. Frontiers in Aging. 2023;4:1–10.
62. Zimmerman SC, Ferguson EL, Choudhary V, et al. Metformin Cessation and Dementia Incidence. JAMA Network Open. 2023;6(10):e2339723. doi:10.1001/jamanetworkopen.2023.39723
63. OHY Y, Suissa S. Metformin and Cancer: solutions to a Real-World Evidence Failure. Diabetes Care. 2023;46(5):904–912. doi:10.2337/dci22-0047
64. Goodwin PJ, Chen BE, Gelmon KA, et al. Effect of metformin vs placebo on invasive disease-free survival in patients with breast cancer: the MA.32 randomized clinical trial. JAMA. 2022;327(20):1963–1973. doi:10.1001/jama.2022.6147
65. Suissa S, Azoulay L. Metformin and the risk of cancer: time-related biases in observational studies. Diabetes Care. 2012;35(12):2665–2673. doi:10.2337/dc12-0788
66. Iudici M, Porcher R, Riveros C, et al. Time-dependent biases in observational studies of comparative effectiveness research in rheumatology. A methodological review. Ann Rheum Dis. 2019;78(4):562–569. doi:10.1136/annrheumdis-2018-214544
67. Luo F, Das A, Chen J, et al. Metformin in patients with and without diabetes: a paradigm shift in cardiovascular disease management. Cardiovasc Diabetol. 2019;18(1):54. doi:10.1186/s12933-019-0860-y
68. Zheng J, Xu M, Yang Q, et al. Efficacy of metformin targets on cardiometabolic health in the general population and non-diabetic individuals: a Mendelian randomization study. EBioMedicine. 2023;96:104803. doi:10.1016/j.ebiom.2023.104803
69. Rena G, Lang CC. Repurposing Metformin for Cardiovascular Disease. Circulation. 2018;137(5):422–424. doi:10.1161/CIRCULATIONAHA.117.031735
70. Fetro C, Scherman D. Drug repurposing in rare diseases: myths and reality. Therapie. 2020;75(2):157–160. doi:10.1016/j.therap.2020.02.006
71. Jacob S, Knoll S, Huhn C, et al. Effects of guanylurea, the transformation product of the antidiabetic drug metformin, on the health of brown trout (Salmo trutta f. fario). PeerJ. 2019;7:e7289. doi:10.7717/peerj.7289
72. Chaignaud P, Gruffaz C, Borreca A, et al. A Methylotrophic Bacterium Growing with the Antidiabetic Drug Metformin as Its Sole Carbon, Nitrogen and Energy Source. Microorganisms. 2022;10(11):2302. doi:10.3390/microorganisms10112302
73. Pentikainen PJ, Neuvonen PJ, Penttila A. Pharmacokinetics of metformin after intravenous and oral administration to man. Eur J Clin Pharmacol. 1979;16(3):195–202. doi:10.1007/BF00562061
74. Tucker GT, Casey C, Phillips PJ, et al. Metformin kinetics in healthy subjects and in patients with diabetes mellitus. Br J Clin Pharmacol. 1981;12(2):235–246. doi:10.1111/j.1365-2125.1981.tb01206.x
75. Christensen MM, Brasch-Andersen C, Green H, et al. The pharmacogenetics of metformin and its impact on plasma metformin steady-state levels and glycosylated hemoglobin A1c. Pharmacogenet Genomics. 2011;21(12):837–850. doi:10.1097/FPC.0b013e32834c0010
76. Graham GG, Punt J, Arora M, et al. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 2011;50(2):81–98. doi:10.2165/11534750-000000000-00000
77. Gong L, Goswami S, Giacomini KM, et al. Metformin pathways: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics. 2012;22(11):820–827. doi:10.1097/FPC.0b013e3283559b22
78. Scheen AJ. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 1996;30(5):359–371. doi:10.2165/00003088-199630050-00003
79. Wang DS, Jonker JW, Kato Y, et al. Involvement of organic cation transporter 1 in hepatic and intestinal distribution of metformin. J Pharmacol Exp Ther. 2002;302(2):510–155. doi:10.1124/jpet.102.034140
80. Takane H, Shikata E, Otsubo K, et al. Polymorphism in human organic cation transporters and metformin action. Pharmacogenomics. 2008;9(4):415–422. doi:10.2217/14622416.9.4.415
81. Liang X, Giacomini KM. Transporters Involved in Metformin Pharmacokinetics and Treatment Response. J Pharm Sci. 2017;106(9):2245–2250. doi:10.1016/j.xphs.2017.04.078
82. Sirtori CR, Franceschini G, Galli-Kienle M, et al. Disposition of metformin (N,N-dimethylbiguanide) in man. Clin Pharmacol Ther. 1978;24(6):683–693. doi:10.1002/cpt1978246683
83. Kajbaf F, De Broe ME, Lalau JD. Therapeutic Concentrations of Metformin: a Systematic Review. Clin Pharmacokinet. 2016;55(4):439–459. doi:10.1007/s40262-015-0323-x
84. Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–1174. doi:10.1172/JCI13505
85. Shaw RJ, Lamia KA, Vasquez D, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310(5754):1642–1646. doi:10.1126/science.1120781
86. Zhou K, Bellenguez C, Spencer CC, et al.; GoDARTS and UKPDS Diabetes Pharmacogenetics Study Group; Wellcome Trust Case Control Consortium 2. Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet. 2011;43(2):117–120. doi:10.1038/ng.735.
87. Menendez JA, Cufí S, Oliveras-Ferraros C, et al. Metformin and the ATM DNA damage response (DDR): accelerating the onset of stress-induced senescence to boost protection against cancer. Aging. 2011;3(11):1063–1077. doi:10.18632/aging.100407
88. Meng S, Cao J, He Q, et al. Metformin activates AMP-activated protein kinase by promoting formation of the αβγ heterotrimeric complex. J Biol Chem. 2015;290(6):3793–3802. doi:10.1074/jbc.M114.604421
89. Evans JM, Donnelly LA, Emslie-Smith AM, et al. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330(7503):1304–1305. doi:10.1136/bmj.38415.708634.F7
90. Yee SW, Chen L, Giacomini KM. The role of ATM in response to metformin treatment and activation of AMPK. Nat Genet. 2012;44(4):359–360. doi:10.1038/ng.2236
91. Potente M, Ghaeni L, Baldessari D, et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007;21(20):2644–2658. doi:10.1101/gad.435107
92. Arunachalam G, Samuel SM, Marei I, et al. Metformin modulates hyperglycaemia-induced endothelial senescence and apoptosis through SIRT1. Br J Pharmacol. 2014;171(2):523–535. doi:10.1111/bph.12496
93. Zhan Y-Y, Chen Y, Zhang Q, et al. The orphan nuclear receptor Nur77 regulates LKB1 localization and activates AMPK. Nature Chemical Biology. 2012;8(11):897–904. doi:10.1038/nchembio.1069
94. Venu VKP, Saifeddine M, Mihara K, et al. Metformin Prevents Hyperglycemia-Associated, Oxidative Stress-Induced Vascular Endothelial Dysfunction: essential Role for the Orphan Nuclear Receptor Human Nuclear Receptor 4A1 (Nur77). Mol Pharmacol. 2021;100(5):428–455. doi:10.1124/molpharm.120.000148
95. El-Mir MY, Nogueira V, Fontaine E, et al. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275(1):223–228. doi:10.1074/jbc.275.1.223
96. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;3.
97. Bridges HR, Blaza JN, Yin Z, et al. Structural basis of mammalian respiratory complex I inhibition by medicinal biguanides. Science. 2023;379(6630):351–357. doi:10.1126/science.ade3332
98. Timmins P, Donahue S, Meeker J, et al. Steady-state pharmacokinetics of a novel extended-release metformin formulation. Clin Pharmacokinet. 2005;44(7):721–729. doi:10.2165/00003088-200544070-00004
99. Wilcock C, Wyre ND, Bailey CJ. Subcellular distribution of metformin in rat liver. J Pharm Pharmacol. 1991;43(6):442–444. doi:10.1111/j.2042-7158.1991.tb03507.x
100. Wilcock C, Bailey CJ. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica. 1994;24(1):49–57. doi:10.3109/00498259409043220
101. Kinaan M, Ding H, Triggle CR. Metformin: an Old Drug for the Treatment of Diabetes but a New Drug for the Protection of the Endothelium. Med Princ Pract. 2015;24(5):401–415. doi:10.1159/000381643
102. He L, Wondisford FE. Metformin action: concentrations matter. Cell Metab. 2015;21(2):159–162. doi:10.1016/j.cmet.2015.01.003
103. Fontaine E. Metformin-Induced Mitochondrial Complex I Inhibition: facts, Uncertainties, and Consequences. Front Endocrinol (Lausanne). 2018;9:753. doi:10.3389/fendo.2018.00753
104. Di Magno L, Di Pastena F, Bordone R, et al. The Mechanism of Action of Biguanides: new Answers to a Complex Question. Cancers (Basel). 2022;14. doi:10.3390/cancers14133220
105. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–262. doi:10.1038/nrm3311
106. Steinberg GR, Hardie DG. New insights into activation and function of the AMPK. Nat Rev Mol Cell Biol. 2023;24:255–272. doi:10.1038/s41580-022-00547-x
107. Cantó C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009;20(2):98–105. doi:10.1097/MOL.0b013e328328d0a4
108. Krausova L, Stejskalova L, Wang H, et al. Metformin suppresses pregnane X receptor (PXR)-regulated transactivation of CYP3A4 gene. Biochem Pharmacol. 2011;82(11):1771–1780. doi:10.1016/j.bcp.2011.08.023
109. McCreight LJ, Bailey CJ, Pearson ER. Metformin and the gastrointestinal tract. Diabetologia. 2016;59(3):426–435. doi:10.1007/s00125-015-3844-9
110. Chen ZP, Mitchelhill KI, Michell BJ, et al. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999;443(3):285–289. doi:10.1016/s0014-5793(98)01705-0
111. Dimmeler S, Fleming I, Fisslthaler B, et al. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399(6736):601–605. doi:10.1038/21224
112. Morrow VA, Foufelle F, Connell JM, et al. Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. J Biol Chem. 2003;278(34):31629–31639. doi:10.1074/jbc.M212831200
113. Calvert JW, Gundewar S, Jha S, et al. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes. 2008;57(3):696–705. doi:10.2337/db07-1098
114. Driver C, Bamitale KDS, Kazi A, et al. Cardioprotective Effects of Metformin. J Cardiovasc Pharmacol. 2018;72(2):121–127. doi:10.1097/FJC.0000000000000599
115. Bailey CJ, Nattrass M. Treatment--metformin. Baillieres Clin Endocrinol Metab. 1988;2(2):455–476. doi:10.1016/s0950-351x(88)80043-0
116. Lkhagva B, Lee TW, Lin YK. Disturbed Cardiac Metabolism Triggers Atrial Arrhythmogenesis in Diabetes Mellitus: energy Substrate Alternate as a Potential Therapeutic Intervention. Cells. 2022;11(18):2915. doi:10.3390/cells11182915
117. Seyed Ahmadi S, Svensson AM, Pivodic A, et al. Risk of atrial fibrillation in persons with type 2 diabetes and the excess risk in relation to glycaemic control and renal function: a Swedish cohort study. Cardiovasc Diabetol. 2020;19(1):9. doi:10.1186/s12933-019-0983-1
118. Tan X, Pan X, Wu X, et al. Glucagon-like peptide-1 receptor agonists as add-on therapy to insulin for type 1 diabetes mellitus. Front Pharmacol. 2023;14:975880. doi:10.3389/fphar.2023.975880
119. Dowling RJO, Niraula S, Stambolic V, et al. Metformin in cancer: translational challenges. J Mol Endocrinol. 2012;48(3):R31–43. doi:10.1530/JME-12-0007
120. Al-Inany H, Johnson N. Drugs for anovulatory infertility in polycystic ovary syndrome. BMJ. 2006;332(7556):1461–1462. doi:10.1136/bmj.332.7556.1461
121. Jakubowicz D, Seppala M. Effects of Metformin on Hyperinsulinemia, Hyperandrogenism and Reproduction in Women with Polycystic Ovarian Syndrome. J Endocrinol Diabetes Obes. 2014;2(2):1034. doi:10.1016/j.jacc.2014.02.555
122. Glueck CJ, Wang P, Kobayashi S, et al. Metformin therapy throughout pregnancy reduces the development of gestational diabetes in women with polycystic ovary syndrome. Fertil Steril. 2002;77(3):520–525. doi:10.1016/s0015-0282(01)03202-2
123. Fougner KJ, Vanky E, Carlsen SM. Metformin has no major effects on glucose homeostasis in pregnant women with PCOS: results of a randomized double-blind study. Scand J Clin Lab Invest. 2008;68(8):771–776. doi:10.1080/00365510802254620
124. Legro RS. Metformin during pregnancy in polycystic ovary syndrome: another vitamin bites the dust. J Clin Endocrinol Metab. 2010;95(12):5199–5202. doi:10.1210/jc.2010-2301
125. Dunne F, Newman C, Alvarez-Iglesias A, et al. Early Metformin in Gestational Diabetes: a Randomized Clinical Trial. JAMA. 2023;330(16):1547–1556. doi:10.1001/jama.2023.19869
126. Wensink MJ, Lu Y, Tian L, et al. Preconception Antidiabetic Drugs in Men and Birth Defects in Offspring: A Nationwide Cohort Study. Ann Intern Med. 2022;175(5):665–673. doi:10.7326/M21-4389
127. Given JE, Loane M, Garne E, et al. Metformin exposure in first trimester of pregnancy and risk of all or specific congenital anomalies: exploratory case-control study. BMJ. 2018;361:k2477. doi:10.1136/bmj.k2477
128. Tarry-Adkins JL, Ozanne SE, Aiken CE. Impact of metformin treatment during pregnancy on maternal outcomes: a systematic review/meta-analysis. Sci Rep. 2021;11(1):9240. doi:10.1038/s41598-021-88650-5
129. Brand KMG, Saarelainen L, Sonajalg J, et al. Metformin in pregnancy and risk of adverse long-term outcomes: a register-based cohort study. BMJ Open Diabetes Res Care. 2022;10(1):e002363. doi:10.1136/bmjdrc-2021-002363
130. Vanky E, Zahlsen K, Spigset O, et al. Placental passage of metformin in women with polycystic ovary syndrome. Fertil Steril. 2005;83(5):1575–1578. doi:10.1016/j.fertnstert.2004.11.051
131. Nguyen L, Chan SY, Teo AKK. Metformin from mother to unborn child - Are there unwarranted effects? EBioMedicine. 2018;35:394–404. doi:10.1016/j.ebiom.2018.08.047
132. Feig DS, Donovan LE, Zinman B, et al. MiTy Collaborative Group. Metformin in women with type 2 diabetes in pregnancy (MiTy): a multicentre, international, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2020:834–844. doi:10.1016/S2213-8587(20)30310-7
133. Wang L, Hou Y, Meng D, et al. Vitamin B12 and Folate Levels During Pregnancy and Risk of Gestational Diabetes Mellitus: a Systematic Review and Meta-Analysis. Front Nutr. 2021;8:670289. doi:10.3389/fnut.2021.670289
134. Lee N, Hebert MF, Wagner DJ, et al. Organic Cation Transporter 3 Facilitates Fetal Exposure to Metformin during Pregnancy. Mol Pharmacol. 2018;94(4):1125–1131. doi:10.1124/mol.118.112482
135. Briggs GG, Ambrose PJ, Nageotte MP, et al. Excretion of metformin into breast milk and the effect on nursing infants. Obstet Gynecol. 2005;105(6):1437–1441. doi:10.1097/01.AOG.0000163249.65810.5b
136. George MM, Copeland KC. Current Treatment Options for Type 2 Diabetes Mellitus in Youth: today’s Realities and Lessons from the TODAY Study. Curr Diab Rep. 2013;13:72–80. doi:10.1007/s11892-012-0334-z
137. Priya G, Kalra S. Metformin in the management of diabetes during pregnancy and lactation. Drugs Context. 2018;7:212523. doi:10.7573/dic.212523
138. Niemuth NJ, Klaper RD. Emerging wastewater contaminant metformin causes intersex and reduced fecundity in fish. Chemosphere. 2015;135:38–45. doi:10.1016/j.chemosphere.2015.03.060
139. Niemuth NJ, Klaper RD. Low-dose metformin exposure causes changes in expression of endocrine disruption-associated genes. Aquat Toxicol. 2018;195:33–40. doi:10.1016/j.aquatox.2017.12.003
140. Alla LNR, Monshi M, Siddiqua Z, et al. Detection of endocrine disrupting chemicals in Danio rerio and Daphnia pulex: step-one, behavioral screen. Chemosphere. 2021;271:129442. doi:10.1016/j.chemosphere.2020.129442
141. Suhre K, Stephan N, Zaghlool S, et al. Matching Drug Metabolites from Non-Targeted Metabolomics to Self-Reported Medication in the Qatar Biobank Study. Metabolites. 2022;12(3):249. doi:10.3390/metabo12030249
142. Petrie JR, Chaturvedi N, Ford I, et al. Metformin in adults with type 1 diabetes: d esign and Methods of REducing with MetfOrmin Vascular Adverse Lesions (REMOVAL): a n international multicentre trial. Diabetes Obesity Metab. 2017;19(4):509–516. doi:10.1111/dom.12840
143. Petrie JR, Chaturvedi N, Ford I, et al. Cardiovascular and metabolic effects of metformin in patients with type 1 diabetes (REMOVAL): a double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017;5(8):597–609. doi:10.1016/S2213-8587(17)30194-8
144. Nasri H, Rafieian-Kopaei M. Metformin: current knowledge. J Res Med Sci. 2014;19(7):658–664.
145. McGovern A, Tippu Z, Hinton W, et al. Comparison of medication adherence and persistence in type 2 diabetes: a systematic review and meta analysis. Diabetes Obesity Metab. 2018;20(4):1040–1043. doi:10.1111/dom.13160
146. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60(9):1577–1585. doi:10.1007/s00125-017-4342-z
147. Bonnet F, Scheen A. Understanding and overcoming metformin gastrointestinal intolerance. Diabetes Obes Metab. 2017;19(4):473–481. doi:10.1111/dom.12854
148. Wong AKF, Howie J, Petrie JR, et al. AMP-activated protein kinase pathway: a potential therapeutic target in cardiometabolic disease. Clin Sci. 2009;116(8):607–620. doi:10.1042/CS20080066
149. Bell DSH. Metformin-induced vitamin B12 deficiency can cause or worsen distal symmetrical, autonomic and cardiac neuropathy in the patient with diabetes. Diabetes Obes Metab. 2022;24(8):1423–1428. doi:10.1111/dom.14734
150. Bauman WA, Shaw S, Jayatilleke E, et al. Increased intake of calcium reverses vitamin B12 malabsorption induced by metformin. Diabetes Care. 2000;23(9):1227–1231. doi:10.2337/diacare.23.9.1227
151. Sayedali E, Yalin AE, Yalin S. Association between metformin and vitamin B12 deficiency in patients with type 2 diabetes. World J Diabetes. 2023;14(5):585–593. doi:10.4239/wjd.v14.i5.585
152. Wåhlén A, Haenni A, Johansson HE. Do we need to measure vitamin B12 and magnesium in morbidly obese patients with type 2 diabetes mellitus? Diabetes Metab Syndr Obes. 2017;10:151–154. doi:10.2147/DMSO.S131340
153. Chowdhury M, Nevitt S, Eleftheriadou A, et al. Cardiac autonomic neuropathy and risk of cardiovascular disease and mortality in type 1 and type 2 diabetes: a meta-analysis. BMJ Open Diabetes Res Care. 2021;9(2):e002480. doi:10.1136/bmjdrc-2021-002480
154. Nam YH, Brensinger CM, Bilker WB, et al. Serious Hypoglycemia and Use of Warfarin in Combination With Sulfonylureas or Metformin. Clin Pharmacol Ther. 2019;105(1):210–218. doi:10.1002/cpt.1146
155. Meadows M. Why drugs get pulled off the market. FDA Consum. 2002;36(1):11–17.
156. Stades AME, Heikens JT, Erkelens DW, et al. Metformin and lactic acidosis: cause or coincidence? A review of case reports. J Int Med. 2004;255(2):179–187. doi:10.1046/j.1365-2796.2003.01271.x
157. Stacpoole PW, Wright EC, Baumgartner TG, et al. Natural history and course of acquired lactic acidosis in adults. DCA-Lactic Acidosis Study Group. Am J Med. 1994;97(1):47–54. doi:10.1016/0002-9343(94)90047-7
158. Flory JH, Hennessy S, Bailey CJ, Inzucchi SE. Reports of Lactic Acidosis Attributed to Metformin, 2015-2018. Diabetes Care. 2020;43(1):244–246. doi:10.2337/dc19-0923
159. Stage TB, Brøsen K, Christensen MMH. A Comprehensive Review of Drug-Drug Interactions with Metformin. Clin Pharmacokinet. 2015;54(8):811–824. doi:10.1007/s40262-015-0270-6.159
160. May M, Schindler C. Clinically and pharmacologically relevant interactions of antidiabetic drugs. Ther Adv Endocrinol Metab. 2016;7(2):69–83. doi:10.1177/2042018816638050
161. Bachmakov I, Glaeser H, Fromm MF, König J. Interaction of oral antidiabetic drugs with hepatic uptake transporters: focus on organic anion transporting polypeptides and organic cation transporter 1. Diabetes. 2008;57(6):1463–1469. doi:10.2337/db07-1515
162. Choi MK, Jin QR, Ahn SH, et al. Sitagliptin attenuates metformin-mediated AMPK phosphorylation through inhibition of organic cation transporters. Xenobiotica. 2010;40(12):817–825. doi:10.3109/00498254.2010.520349
163. Leon BM, Maddox TM. Diabetes and cardiovascular disease: epidemiology, biological mechanisms, treatment recommendations and future research. World J Diabetes. 2015;6(13):1246–1258. doi:10.4239/wjd.v6.i13.1246
164. Deshmukh A, Ghannam M, Liang J, et al. Effect of metformin on outcomes of catheter ablation for atrial fibrillation. J Cardiovasc Electrophysiol. 2021;32(5):1232–1239. doi:10.1111/jce.14954
165. Stoll L, Lo JC. GLP-1 Receptor Agonists, the Holy Grail Preventing Atrial Fibrillation in Patients With T2D? JACC Basic Transl Sci. 2023;8(8):937–938. doi:10.1016/j.jacbts.2023.03.022
166. Bohne LJ, Jansen HJ, Dorey TW, et al. Glucagon-Like Peptide-1 Protects Against Atrial Fibrillation and Atrial Remodeling in Type 2 Diabetic Mice. JACC Basic Transl Sci. 2023;8(8):922–936. doi:10.1016/j.jacbts.2023.01.005
167. Bu Y, Peng M, Tang X, et al. Protective effects of metformin in various cardiovascular diseases: clinical evidence and AMPK-dependent mechanisms. J Cell Mol Med. 2022;26(19):4886–4903. doi:10.1111/jcmm.17519
168. Raubenheimer PJ, Cushman WC, Avezum A, et al. Dulaglutide and incident atrial fibrillation or flutter in patients with type 2 diabetes: a post hoc analysis from the REWIND randomized trial. Diabetes Obes Metab. 2022;24(4):704–712. doi:10.1111/dom.14634
169. Wakili R, Voigt N, Kääb S, et al. Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Invest. 2011;121(8):2955–2968. doi:10.1172/JCI46315
170. Schotten U, Verheule S, Kirchhof P, et al. Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol Rev. 2011;91(1):265–325. doi:10.1152/physrev.00031.2009
171. Yang F, Liu HH, Zhang L, et al. Advanced Glycation End Products Downregulate Connexin 43 and Connexin 40 in Diabetic Atrial Myocytes via the AMPK Pathway. Diabetes Metab Syndr Obes. 2023;16:3045–3056. doi:10.2147/DMSO.S419189
172. Nabauer M, Gerth A, Limbourg T, et al. The Registry of the German Competence NETwork on Atrial Fibrillation: patient characteristics and initial management. Europace. 2009;11(4):423–434. doi:10.1093/europace/eun369
173. Goudis CA, Korantzopoulos P, Ntalas IV, et al. Diabetes mellitus and atrial fibrillation: pathophysiological mechanisms and potential upstream therapies. Int J Cardiol. 2015;184(1):617–622.
174. Michaud GF, Stevenson WG. Atrial Fibrillation. N Engl J Med. 2021;384(4):353–361. doi:10.1056/NEJMcp2023658
175. Li J, Gao M, Zhang M, et al. Treatment of atrial fibrillation: a comprehensive review and practice guide. Cardiovasc J Afr. 2020;31(3):153–158. doi:10.5830/CVJA-2019-064
176. Aktas G, Atak Tel BM, et al. Treatment of type 2 diabetes patients with heart conditions. Expert Rev Endocrinol Metab. 2023;18(3):255–265. doi:10.1080/17446651.2023.2204941
177. Qian LL, Liu XY, Li XY, et al. Effects of Electrical Remodeling on Atrial Fibrillation in Diabetes Mellitus. Rev Cardiovasc Med. 2023;24(1):3.
178. Snell-Bergeon JK, Wadwa RP. Hypoglycemia, Diabetes, and Cardiovascular Disease. Diabetes Technol Ther. 2012;14 Suppl 1(Suppl 1):S51–8. doi:10.1089/dia.2012.0031
179. Tsujimoto T, Sugiyama T, Noda M, et al. Intensive Glycemic Therapy in Patients With Type 2 Diabetes on β-Blockers. Diabetes Care. 2016;39(10):1818–1826. doi:10.2337/dc16-0721
180. Yu O, Azoulay L, Yin H, et al. Sulfonylureas as Initial Treatment for Type 2 Diabetes and the Risk of Severe Hypoglycemia. Am J Med. 2017;131(3):317.e11–317.e22. doi:10.1016/j.amjmed.2017.09.044
181. Akirov A, Grossman A, Shochat T, Shimon I. Hyperglycemia on admission and hospitalization outcomes in patients with atrial fibrillation. Clin Cardiol. 2017;40(11):1123–1128. doi:10.1002/clc.22801
182. Yuan M, Gong M, Zhang Z, et al. Hyperglycemia Induces Endoplasmic Reticulum Stress in Atrial Cardiomyocytes, and Mitofusin-2 Downregulation Prevents Mitochondrial Dysfunction and Subsequent Cell Death. Oxid Med Cell Longev. 2020:6569728. doi:10.1155/2020/6569728
183. Liu C, Fu H, Li J, et al. Hyperglycemia aggravates atrial interstitial fibrosis, ionic remodeling and vulnerability to atrial fibrillation in diabetic rabbits. Anadolu Kardiyol Derg. 2012;12(7):543–550. doi:10.5152/akd.2012.188
184. Mujović N, Marinković M, Mihajlović M, et al. Risk factor modification for the primary and secondary prevention of atrial fibrillation. Part 1. Kardiol Pol. 2020;78(3):192–202. doi:10.33963/KP.15240
185. Ceriello A, Monnier L, Owens D. Glycaemic variability in diabetes: clinical and therapeutic implications. Lancet Diabetes Endocrinol. 2019;7(3):221–230. doi:10.1016/S2213-8587(18)30136-0
186. Singh R, Barden A, Mori T, et al. Advanced glycation end-products: a review. Diabetologia. 2002;44(2):129–146. doi:10.1007/s001250051591
187. Zheng DL, Wu QR, Zeng P, et al. Advanced glycation end products induce senescence of atrial myocytes and increase susceptibility of atrial fibrillation in diabetic mice. Aging Cell. 2022;21(12):e13734. doi:10.1111/acel.13734
188. Raposeiras-Roubín S, Rodiño-Janeiro BK, Grigorian-Shamagian L, et al. Evidence for a role of advanced glycation end products in atrial fibrillation. Int J Cardiol. 2012;157(3):397–402. doi:10.1016/j.ijcard.2011.05.072
189. Lee TW, Lee TI, Lin YK, et al. Effect of antidiabetic drugs on the risk of atrial fibrillation: mechanistic insights from clinical evidence and translational studies. Cell Mol Life Sci. 2021;78:923–934. doi:10.1007/s00018-020-03648-y
190. Fu H. Products (AGEs) and their Receptor Axis in Atrial Fibrillation. Mini Rev Med Chem. 2019;19(13):1040–1048. doi:10.2174/1389557519666190311140737
191. Roth GA. Factors, 1990–2019: update From the GBD 2019 Study. J Am Coll Cardiol. 2020;76(25):2982. doi:10.1016/j.jacc.2020.11.010
192. Mather KJ, Verma S, Anderson TJ. Improved endothelial function with metformin in type 2 diabetes mellitus. J Am Coll Cardiol. 2001;37(5):1344–1350. doi:10.1016/s0735-1097(01)01129-9
193. Triggle CR, Ding H. Metformin is not just an antihyperglycaemic drug but also has protective effects on the vascular endothelium. Acta Physiol (Oxf). 2017;219(1):138–151. doi:10.1111/apha.12644
194. Hong J, Zhang Y, Lai S, et al. Effects of Metformin Versus Glipizide on Cardiovascular Outcomes in Patients With Type 2 Diabetes and Coronary Artery Disease. Diabetes Care. 2013;36(5):1304–1311. doi:10.2337/dc12-0719
195. Vaccaro O, Masulli M, Nicolucci A, et al. Effects on the incidence of cardiovascular events of the addition of pioglitazone versus sulfonylureas in patients with type 2 diabetes inadequately controlled with metformin (TOSCA.IT): a randomised, multicentre trial. Lancet Diabetes Endocrinol. 2017;5(11):887–987. doi:10.1016/S2213-8587(17)30317-0
196. Paiva M, Riksen NP, Davidson SM, et al. Metformin prevents myocardial reperfusion injury by activating the adenosine receptor. J Cardiovasc Pharmacol. 2009;53(5):373–378. doi:10.1097/FJC.0b013e31819fd4e7
197. Riksen NP, Rongen GA. Targeting adenosine receptors in the development of cardiovascular therapeutics. Expert Rev Clin Pharmacol. 2012;5(2):199–218. doi:10.1586/ecp.12.8
198. Rankin AC, Brooks R, Ruskin JN, et al. Adenosine and the treatment of supraventricular tachycardia. Am J Med. 1992;92(6):655–664. doi:10.1016/0002-9343(92)90784-9
199. Koeppen M, Eckle T, Eltzschig HK. Selective deletion of the A1 adenosine receptor abolishes heart-rate slowing effects of intravascular adenosine in vivo. PLoS One. 2009;4(8):e6784. doi:10.1371/journal.pone.0006784
200. Mas M. A Close Look at the Endothelium: its Role in the Regulation of Vasomotor Tone. Eur Urol Suppl. 2009;8(2):48–57. doi:10.1016/j.eursup.2008.10.011
201. Domeier TL, Segal SS. Electromechanical and pharmacomechanical signalling pathways for conducted vasodilatation along endothelium of hamster feed arteries. J Physiol. 2007;579(Pt 1):175–186. doi:10.1113/jphysiol.2006.124529
202. Dora KA, Lin J, Borysova L, et al. Signaling and structures underpinning conducted vasodilation in human and porcine intramyocardial coronary arteries. Front Cardiovasc Med. 2022;9:980628. doi:10.3389/fcvm.2022.980628
203. Asagami T, Abbasi F, Stuelinger M, et al. Metformin treatment lowers asymmetric dimethylarginine concentrations in patients with type 2 diabetes. Metabolism. 2002;51(7):843–846. doi:10.1053/meta.2002.33349
204. Tsai CM, Kuo HC. Hsu et al Metformin reduces asymmetric dimethylarginine and prevents hypertension in spontaneously hypertensive rats. Transl Res. 2014;164(6):452–459. doi:10.1016/j.trsl.2014.07.005
205. Takahashi N, Shibata R, Ouchi N, et al. Metformin stimulates ischemia-induced revascularization through an eNOS dependent pathway in the ischemic hindlimb mice model. J Vasc Surg. 2015;61(2):489–496. doi:10.1016/j.jvs.2013.09.061
206. Ding Y, Zhou Y, Ling P, et al. Metformin in cardiovascular diabetology: a focused review of its impact on endothelial function. Theranostics. 2021;11(19):9376–9396. doi:10.7150/thno.64706
207. Triposkiadis F, Xanthopoulos A, Parissis J, et al. Pathogenesis of chronic heart failure: cardiovascular aging, risk factors, comorbidities, and disease modifiers. Heart Fail Rev. 2022;27:337–344. doi:10.1007/s10741-020-09987-z
208. Haghbin H, Gangwani MK, Ravi SJK, et al. Nonalcoholic fatty liver disease and atrial fibrillation: possible pathophysiological links and therapeutic interventions. Ann Gastroenterol. 2020;33(6):603–614. doi:10.20524/aog.2020.0550
209. Kim HS, Ren G, Kim T, et al. Metformin reduces saturated fatty acid-induced lipid accumulation and inflammatory response by restoration of autophagic flux in endothelial cells. Sci Rep. 2020;10:13523. doi:10.1038/s41598-020-70347-w
210. Kim JA, Montagnani M, Chandrasekran S, Quon MJ. Role of lipotoxicity in endothelial dysfunction. Heart Fail Clin. 2012;8(4):589–607. doi:10.1016/j.hfc.2012.06.012
211. Lee TI, Kao YH, Chen YC, et al. Cardiac metabolism, inflammation, and peroxisome proliferator-activated receptors modulated by 1,25-dihydroxyvitamin D3 in diabetic rats. Int J Cardiol. 2014;176(1):151–157. doi:10.1016/j.diabres.2013.01.008
212. Mellbin LG, Malmberg K, Norhammar A, et al. The impact of glucose lowering treatment on long-term prognosis in patients with type 2 diabetes and myocardial infarction: a report from the DIGAMI 2 trial. Eur Heart J. 2008;29(2):166–176. doi:10.1093/eurheartj/ehm518
213. Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res. 2013;112(8):1171–1188. doi:10.1161/CIRCRESAHA.111.300233
214. Li C, Reif MM, Craige SM, et al. Endothelial AMPK activation induces mitochondrial biogenesis and stress adaptation via eNOS-dependent mTORC1 signaling. Nitric Oxide. 2016;55-56:45–53. doi:10.1016/j.niox.2016.03.003
215. Halling JF, Pilegaard H. PGC-1α-mediated regulation of mitochondrial function and physiological implications. Appl Physiol Nutr Metab. 2020;45(9):927–936. doi:10.1139/apnm-2020-0005
216. Khan AA, Thomas GN, Lip GYH, Shantsila A. Endothelial function in patients with atrial fibrillation. Ann Med. 2020;52(1–2):1–11. doi:10.1080/07853890.2019.1711158
217. Freedman JE, Loscalzo J, Barnard MR, et al. Nitric oxide released from activated platelets inhibits platelet recruitment. J Clin Invest. 1997;100(2):350–356. doi:10.1172/JCI119540
218. Endemann DH, Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol. 2004;15(8):1983–1992. doi:10.1097/01.ASN.0000132474.50966.DA
219. Cai H, Li Z, Goette A, et al. Downregulation of endocardial nitric oxide synthase expression and nitric oxide production in atrial fibrillation: potential mechanisms for atrial thrombosis and stroke. Circulation. 2002;106(22):2854–2858. doi:10.1161/01.cir.0000039327.11661.16
220. Ersoy C, Kiyici S, Budak F, et al. The effect of metformin treatment on VEGF and PAI-1 levels in obese type 2 diabetic patients. Diabet Res Clin Pract. 2008;81(1):56–60. doi:10.1016/j.diabres.2008.02.006
221. Carnes CA, Chung MK. Nakayama T, et al Ascorbate attenuates atrial pacing-induced peroxynitrite formation and electrical remodeling and decreases the incidence of postoperative atrial fibrillation. Circ Res. 2001;89(6):E32–8. doi:10.1161/hh1801.097644
222. Lip GY, Blann A. von Willebrand factor: a marker of endothelial dysfunction in vascular disorders? Cardiovasc Res. 1997;34(2):255–265. doi:10.1016/s0008-6363(97)00039-4
223. Conway DS, Pearce LA, Chin BS, et al. Prognostic value of plasma von Willebrand factor and soluble P-selectin as indices of endothelial damage and platelet activation in 994 patients with nonvalvular atrial fibrillation. Circulation. 2003;107(25):3141–3145. doi:10.1161/01.CIR.0000077912.12202.FC
224. Spiel AO, Gilbert JC, Jilma B. von Willebrand factor in cardiovascular disease: focus on acute coronary syndromes. Circulation. 2008;117(11):1449–1459. doi:10.1161/CIRCULATIONAHA.107.722827
225. Bell DSH, Goncalves E. Stroke in the patient with diabetes (Part 2) – prevention and the effects of glucose lowering therapies. Diabet Res Clin Pract. 2020;164. doi:10.1016/j.diabres.2020.108199
226. Saez JC, Berthoud VM, Branes MC, et al. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83(4):1359–1400. doi:10.1152/physrev.00007.2003
227. Rodríguez-Sinovas A, Sánchez JA, Valls-Lacalle L, et al. Connexins in the Heart: regulation, Function and Involvement in Cardiac Disease. Int J Mol Sci. 2021;22(9):4413. doi:10.3390/ijms22094413
228. Kelm NQ, Solinger JC, Piell KM, Cole MP. Conjugated Linoleic Acid-Mediated Connexin-43 Remodeling and Sudden Arrhythmic Death in Myocardial Infarction. Int J Mol Sci. 2023;24(13):11208. doi:10.3390/ijms241311208
229. Severs NJ, Dupont E, Coppen SR, et al. Remodelling of gap junctions and connexin expression in heart disease. Biochim Biophys Acta. 2004;1662(1–2):138–148. doi:10.1016/j.bbamem.2003.10.019
230. Solan JL, Lampe PD. Specific Cx43 phosphorylation events regulate gap junction turnover in vivo. FEBS Lett. 2014;588(8):1423–1429. doi:10.1016/j.febslet.2014.01.049
231. Liu D, He J, Zhou D, et al. Connexin43 Overexpression Exacerbates Myocardial Ischemic Reperfusion Injury in Diabetes via Modulating Cardiac Autophagy. FASEB J. 2022;36(S1):7849. doi:10.1096/fasebj.2022.36.S1.L7849
232. Ek-Vitorín JF, Pontifex TK, Burt JM. Cx43 Channel Gating and Permeation: multiple Phosphorylation-Dependent Roles of the Carboxyl Terminus. Int J Mol Sci. 2018;19(6):1659. doi:10.3390/ijms19061659
233. Remo BF, Qu J, Volpicelli FM, et al. Phosphatase-resistant gap junctions inhibit pathological remodeling and prevent arrhythmias. Circ Res. 2011;108(12):1459–1466. doi:10.1161/CIRCRESAHA.111.244046
234. Ozcan C, Battaglia E, Young R, et al. LKB1 knockout mouse develops spontaneous atrial fibrillation and provides mechanistic insights into human disease process. J Am Heart Assoc. 2015;4(3):e001733. doi:10.1161/JAHA.114.001733
235. Ozcan C, Li Z, Kim G, et al. Molecular Mechanism of the Association Between Atrial Fibrillation and Heart Failure Includes Energy Metabolic Dysregulation Due to Mitochondrial Dysfunction. J Card Fail. 2019;25(11):911–920. doi:10.1016/j.cardfail.2019.08.005
236. Ozcan C, Dixit G, Li Z. Activation of AMP-Activated Protein Kinases Prevents Atrial Fibrillation. J Cardiovasc Transl Res. 2021;14(3):492–502. doi:10.1007/s12265-020-10069-6
237. Akoyev V, Takemoto DJ. ZO-1 is required for protein kinase C gamma-driven disassembly of connexin 43. Cell Signal. 2007;19(5):958–967. doi:10.1016/j.cellsig.2006.11.007
238. Wagner LM, Saleh SM, Boyle DJ, et al. Effect of protein kinase Cgamma on gap junction disassembly in lens epithelial cells and retinal cells in culture. Mol Vis. 2002;8:59–66.
239. Gong Y, Wang C, Jiang Y, et al. Metformin Inhibits Tumor Metastasis through Suppressing Hsp90α Secretion in an AMPKα1-PKCγ Dependent Manner. Cells. 2020;9(1):144. doi:10.3390/cells9010144
240. Rhett JM, Ongstad EL, Jourdan J, et al. Cx43 Associates with Nav1.5 in the Cardiomyocyte Perinexus. J Membr Biol. 2012;245(7):411–422. doi:10.1007/s00232-012-9465-z
241. Kim GE, Ross JL, Xie C, et al. LKB1 deletion causes early changes in atrial channel expression and electrophysiology prior to atrial fibrillation. Cardiovasc Res. 2015;108(1):197–208. doi:10.1093/cvr/cvv212
242. Molaei A, Molaei E, Sadeghnia H, et al. LKB1: an emerging therapeutic target for cardiovascular diseases. Life Sci. 2022;306:120844. doi:10.1016/j.lfs.2022.120844
243. Ndembe G, Intini I, Perin E, et al. LKB1: can We Target an Hidden Target? Focus on NSCLC. Front Oncol. 2022;12:889826. doi:10.3389/fonc.2022.889826
244. Pool L, Wijdeveld LFJM, de Groot F, et al. The Role of Mitochondrial Dysfunction in Atrial Fibrillation: translation to Druggable Target and Biomarker Discovery. Int J Mol Sci. 2021;22(16):8463. doi:10.3390/ijms22168463
245. Chen Q, Thompson J, Hu Y, Lesnefsky EJ. Chronic metformin treatment decreases cardiac injury during ischemia-reperfusion by attenuating endoplasmic reticulum stress with improved mitochondrial function. Aging. 2021;13(6):7828–7845. doi:10.18632/aging.202858
246. Sun D, Yang F. Metformin improves cardiac function in mice with heart failure after myocardial infarction by regulating mitochondrial energy metabolism. Biochem Biophys Res Commun. 2017;486(2):329–335. doi:10.1016/j.bbrc.2017.03.036
247. Larsen AH, Jessen N, Nørrelund H, et al. A randomised, double-blind, placebo-controlled trial of metformin on myocardial efficiency in insulin-resistant chronic heart failure patients without diabetes. Eur J Heart Fail. 2020;22(9):1628–1637. doi:10.1002/ejhf.1656
248. Han Y, Xie H, Liu Y, et al. Effect of metformin on all-cause and cardiovascular mortality in patients with coronary artery diseases: a systematic review and an updated meta-analysis. Cardiovasc Diabetol. 2019;18:96. doi:10.1186/s12933-019-0900-7
249. Karnewar S, Neeli PK, Panuganti D, et al. Metformin regulates mitochondrial biogenesis and senescence through AMPK mediated H3K79 methylation: relevance in age-associated vascular dysfunction. Biochim Biophys Acta Mol Basis Dis. 2018;1864(4 Pt A):1115–1128. doi:10.1016/j.bbadis.2018.01.018
250. de Marañón AM, Díaz-Pozo P, Canet F, et al. Metformin modulates mitochondrial function and mitophagy in peripheral blood mononuclear cells from type 2 diabetic patients. Redox Biol. 2022;53:102342. doi:10.1016/j.redox.2022.102342
251. Aatsinki SM, Buler M, Salomäki H, et al. Metformin induces PGC-1α expression and selectively affects hepatic PGC-1α functions. Br J Pharmacol. 2014;171(9):2351–2363. doi:10.1111/bph.12585
252. Targher G, Byrne CD, Tilg H. NAFLD and increased risk of cardiovascular disease: clinical associations, pathophysiological mechanisms and pharmacological implications. Gut. 2020;69(9):1691–1705. doi:10.1136/gutjnl-2020-320622
253. Bai F, Liu Y, Tu T, et al. Metformin regulates lipid metabolism in a canine model of atrial fibrillation through AMPK/PPAR-α/VLCAD pathway. Lipids Health Dis. 2019;18(1):1–9. doi:10.1186/s12944-019-1059-7
254. Zhao W, Zhou L, Novák P, et al. Metabolic Dysfunction in the Regulation of the NLRP3 Inflammasome Activation: a Potential Target for Diabetic Nephropathy. J Diabetes Res. 2022;2022:2193768. doi:10.1155/2022/2193768
255. Yao C, Veleva T, Scott L, et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation. 2018;138(20):2227–2242. doi:10.1161/CIRCULATIONAHA.118.035202
256. Yang F, Qin Y, Wang Y, et al. Metformin Inhibits the NLRP3 Inflammasome via AMPK/mTOR-dependent Effects in Diabetic Cardiomyopathy. Int J Biol Sci. 2019;15(5):1010–1019. doi:10.7150/ijbs.29680
257. Russo I, Frangogiannis NG. Diabetes-associated cardiac fibrosis: cellular effectors, molecular mechanisms and therapeutic opportunities. J Mol Cell Cardiol. 2016;90:84–93. doi:10.1016/j.yjmcc.2015.12.011
258. Zhang Q, Liu T, Ng CY, et al. Diabetes mellitus and atrial remodeling: mechanisms and potential upstream therapies. Cardiovasc Ther. 2014;32(5):233–241. doi:10.1111/1755-5922.12089
259. Jansen HJ, Bohne LJ, Gillis AM, Rose RA. Atrial remodeling and atrial fibrillation in acquired forms of cardiovascular disease. Heart Rhythm O2. 2020;1(2):147–159. doi:10.1016/j.hroo.2020.05.002
260. Yildiz M, Lavie CJ, Morin DP, et al. The complex interplay between diabetes mellitus and atrial fibrillation. Expert Rev Cardiovasc Ther. 2022;20(9):707–717. doi:10.1080/14779072.2022.2115357
261. Ozturk N, Uslu S, Ozdemir S. Diabetes-induced changes in cardiac voltage-gated ion channels. World J Diabetes. 2021;12(1):1–18. doi:10.4239/wjd.v12.i1.1
262. Liu CH, Hua N, Fu X, et al. Metformin regulates atrial SK2 and SK3 expression through inhibiting the PKC/ERK signaling pathway in type 2 diabetic rats. BMC Cardiovasc Disord. 2018;18(1):236. doi:10.1186/s12872-018-0950-x
263. Liu C, Hua N, Fu X, et al. Metformin Regulates the Expression of SK2 and SK3 in the Atria of Rats With Type 2 Diabetes Mellitus Through the NOX4/p38MAPK Signaling Pathway. J Cardiovasc Pharmacol. 2018;72(5):205–213. doi:10.1097/FJC.0000000000000615
264. Jichitu A, Bungau S, Stanescu AMA, et al. Non-Alcoholic Fatty Liver Disease and Cardiovascular Comorbidities: pathophysiological Links, Diagnosis, and Therapeutic Management. Diagnostics. 2021;1(4):689. doi:10.3390/diagnostics11040689
265. Saddik M, Lopaschuk GD. Myocardial triglyceride turnover and contribution to energy substrate utilization in isolated working rat hearts. J Biol Chem. 1991;266(13):8162–8170.
266. Wisneski JA, Stanley WC, Neese RA, et al. Effects of acute hyperglycemia on myocardial glycolytic activity in humans. J Clin Invest. 1990;85(5):1648–1656. doi:10.1172/JCI114616
267. Lopaschuk GD, Ussher JR, Folmes CD, et al. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90(1):207–258. doi:10.1152/physrev.00015.2009
268. Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J Inherit Metab Dis. 2010;33(5):469–477. doi:10.1007/s10545-010-9061-2
269. Greenwell AA, Gopal K, Ussher JR. Myocardial Energy Metabolism in Non-ischemic Cardiomyopathy. Front Physiol. 2020;11:570421. doi:10.3389/fphys.2020.570421
270. Cotter DG, Schugar RC, Crawford PA. Ketone body metabolism and cardiovascular disease. Am J Physiol Heart Circ Physiol. 2013;304(8):H1060–1076. doi:10.1152/ajpheart.00646.2012
271. Ho KL, Zhang L, Wagg C, et al. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc Res. 2019;115(11):1606–1616. doi:10.1093/cvr/cvz045
272. Schugar RC, Moll AR. André d’Avignon D, et al Cardiomyocyte-specific deficiency of ketone body metabolism promotes accelerated pathological remodeling. Mol Metab. 2014;3(7):754–769. doi:10.1016/j.molmet.2014.07.010
273. Barth AS, Merk S, Arnoldi E, et al. Reprogramming of the human atrial transcriptome in permanent atrial fibrillation: expression of a ventricular-like genomic signature. Circ Res. 2005;96(9):1022–1029. doi:10.1161/01.RES.0000165480.82737.33
274. Shingu Y, Yokota T, Takada S, et al. Decreased gene expression of fatty acid binding protein 3 in the atrium of patients with new onset of atrial fibrillation in cardiac perioperative phase. J Cardiol. 2018;71(1):65–70. doi:10.1016/j.jjcc.2017.07.003
275. Shingu Y, Takada S, Yokota T, et al. Correlation between increased atrial expression of genes related to fatty acid metabolism and autophagy in patients with chronic atrial fibrillation. PLoS One. 2020;15(4):e0224713. doi:10.1371/journal.pone.0224713
276. Khawaja O, Bartz TM, Ix JH, et al. Plasma free fatty acids and risk of atrial fibrillation (from the Cardiovascular Health Study). Am J Cardiol. 2012;110(2):212–216. doi:10.1016/j.amjcard.2012.03.010
277. Hansen PS, Clarke RJ, Buhagiar KA, et al. Alloxan-induced diabetes reduces sarcolemmal Na+-K+ pump function in rabbit ventricular myocytes. Am J Physiol Cell Physiol. 2007;292(3):C1070–7. doi:10.1152/ajpcell.00288.2006
278. Barth AS, Tomaselli GF. Cardiac metabolism and arrhythmias. Circ Arrhythm Electrophysiol. 2009;2(3):327–335. doi:10.1161/CIRCEP.108.817320
279. Bueno-Orovio A, Sánchez C, Pueyo E, Rodriguez B. Na/K pump regulation of cardiac repolarization: insights from a systems biology approach. Pflugers Arch. 2014;466(2):183–193. doi:10.1007/s00424-013-1293-1
280. Lenski M, Schleider G, Kohlhaas M, et al. Arrhythmia causes lipid accumulation and reduced glucose uptake. Basic Res Cardiol. 2015;110(4):40. doi:10.1007/s00395-015
281. Dai DF, Chen T, Johnson SC, et al. Cardiac aging: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal. 2012;16(12):1492–1526. doi:10.1089/ars.2011.4179
282. van Rosendael AR, Dimitriu-Leen AC, van Rosendael PJ, et al. Association Between Posterior Left Atrial Adipose Tissue Mass and Atrial Fibrillation. Circ Arrhythm Electrophysiol. 2017;10(2):e004614. doi:10.1161/CIRCEP.116.004614
283. Sutanto H, Lyon A, Lumens J, et al. Cardiomyocyte calcium handling in health and disease: insights from in vitro and in silico studies. Prog Biophys Mol Biol. 2020;157:54–75. doi:10.1016/j.pbiomolbio.2020.02.008
284. Liu GZ, Hou TT, Yuan Y, et al. Fenofibrate inhibits atrial metabolic remodelling in atrial fibrillation through PPAR-α/sirtuin 1/PGC-1α pathway. Br J Pharmacol. 2016;173(6):1095–1109. doi:10.1111/bph.13438
285. Ludhwani D, Wieters JS. Paroxysmal Atrial Fibrillation; 2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK535439/.
286. Liu Y, Bai F, Liu N, et al. The Warburg effect: a new insight into atrial fibrillation. Clin Chim Acta. 2019;499:4–12. doi:10.1016/j.cca.2019.08.029
287. Hiraoka M, Okamoto Y, Sano T. Electrophysiological effects of lactates in mammalian ventricular tissues. J Electrocardiol. 1981;14(1):13–20. doi:10.1016/s0022-0736(81)80023-4
288. Saman S, Opie LH. Mechanism of reduction of action potential duration of ventricular myocardium by exogenous lactate. J Mol Cell Cardiol. 1984;16(7):659–662. doi:10.1016/s0022-2828(84)80629-x
289. Wu P, Zhu T, Huang Y, et al. Current understanding of the contribution of lactate to the cardiovascular system and its therapeutic relevance. Front Endocrinol (Lausanne). 2023;14:1205442. doi:10.3389/fendo.2023.1205442
290. Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485(7400):661–665. doi:10.1038/nature11066
291. Muzurović EM, Vujošević S, Mikhailidis DP. Can We Decrease Epicardial and Pericardial Fat in Patients With Diabetes? J Cardiovasc Pharmacol Ther. 2021;26(5):415–436. doi:10.1177/10742484211006997
292. Su JR, Lu ZH, Su Y, et al. Relationship of Serum Adiponectin Levels and Metformin Therapy in Patients with Type 2 Diabetes. Horm Metab Res. 2016;48(2):92–98. doi:10.1055/s-0035-1569287
293. Macheret F, Bartz TM, Djousse L, et al. Higher circulating adiponectin levels are associated with increased risk of atrial fibrillation in older adults. Heart. 2015;101(17):1368–1374. doi:10.1136/heartjnl-2014-307015
294. El Messaoudi S, Nederlof R, Zuurbier CJ, et al. Effect of metformin pretreatment on myocardial injury during coronary artery bypass surgery in patients without diabetes (MetCAB): a double-blind, randomised controlled trial. Lancet Diabetes Endocrinol. 2015;2(8):615–623. doi:10.1016/S2213-8587(15)00121-7
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.