")
Back to Journals » Journal of Inflammation Research » Volume 17
S100A9 as a Key Myocardial Injury Factor Interacting with ATP5 Exacerbates Mitochondrial Dysfunction and Oxidative Stress in Sepsis-Induced Cardiomyopathy
Authors Pei H , Qu J, Chen J, Zhao G, Lu Z
Received 30 December 2023
Accepted for publication 22 June 2024
Published 9 July 2024 Volume 2024:17 Pages 4483—4503
DOI https://doi.org/10.2147/JIR.S457340
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tara Strutt
Hui Pei,1 Jie Qu,1 Jianming Chen,1 Guangju Zhao,1 ZhongQiu Lu1,2
1Emergency Department, the First Affiliated Hospital of Wenzhou Medical University, Wenzhou, People’s Republic of China; 2Wenzhou Key Laboratory of Emergency and Disaster Medicine, Wenzhou, People’s Republic of China
Correspondence: ZhongQiu Lu, Email [email protected]
Purpose: Sepsis-induced cardiomyopathy (SICM) is a prevalent cardiac dysfunction caused by sepsis. Mitochondrial dysfunction is a crucial pathogenic factor associated with adverse cardiovascular adverse events; however, research on SICM remains insufficient.
Methods: To investigate the factors contributing to the pathological progression of SICM, we performed a comprehensive analysis of transcriptomic data from the GEO database using bioinformatics and machine learning techniques. CRISPR-Cas9 S100A9 knockout mice and primary cardiomyocytes were exposed to lipopolysaccharide to simulate SICM. Transcriptome analysis and mass spectrometry of primary cardiomyocytes were used to determine the potential pathogenic mechanisms of S100A9. The mitochondrial ultrastructure and mitochondrial membrane potential (MMP) were detected using transmission electron microscopy and flow cytometry, respectively. Pink1/Parkin and Drp1 proteins were detected using Western blotting to evaluate mitochondrial autophagy and division. The mtDNA and mRNA levels of mitochondrial transcription factors and synthases were evaluated using real-time polymerase chain reaction.
Results: Bioinformatics analysis identified 12 common differentially expressed genes, including SERPINA3N, LCN2, MS4A6D, LRG1, OSMR, SOCS3, FCGR2b, S100A9, S100A8, CASP4, ABCA8A, and NFKBIZ. Significant S100A9 upregulation was closely associated with myocardial injury exacerbation and cardiac function deterioration. GSEA revealed that myocardial contractile function, oxidative stress, and mitochondrial function were significantly affected by S100A9. Knocking out S100A9 alleviates the inflammatory response and mitochondrial dysfunction. The interaction of S100A9 with ATP5 enhanced mitochondrial division and autophagy, inhibited MMP and ATP synthesis, and induced oxidative stress, which are related to the Nlrp3-Nfkb-Caspase1 and Drp1-Pink1-Parkin signaling pathways. The expression of mitochondrial transcription factors (TFAM and TFBM) and ATP synthetases (ATP6 and ATP8, as well as COX1, COX2, and COX3) was further suppressed by S100A9 in SICM. Targeted S100A9 inhibition by paquinimod partially reversed myocardial mitochondrial dysfunction and oxidative stress.
Conclusion: The interaction of S100A9 with ATP5 exacerbates myocardial damage in sepsis by inducing mitochondrial dysfunction and oxidative stress.
Keywords: S100A9, SICM, bioinformatics analysis, mitochondrial dysfunction, ATP5
Corrigendum for this paper has been published.
Graphical Abstract:
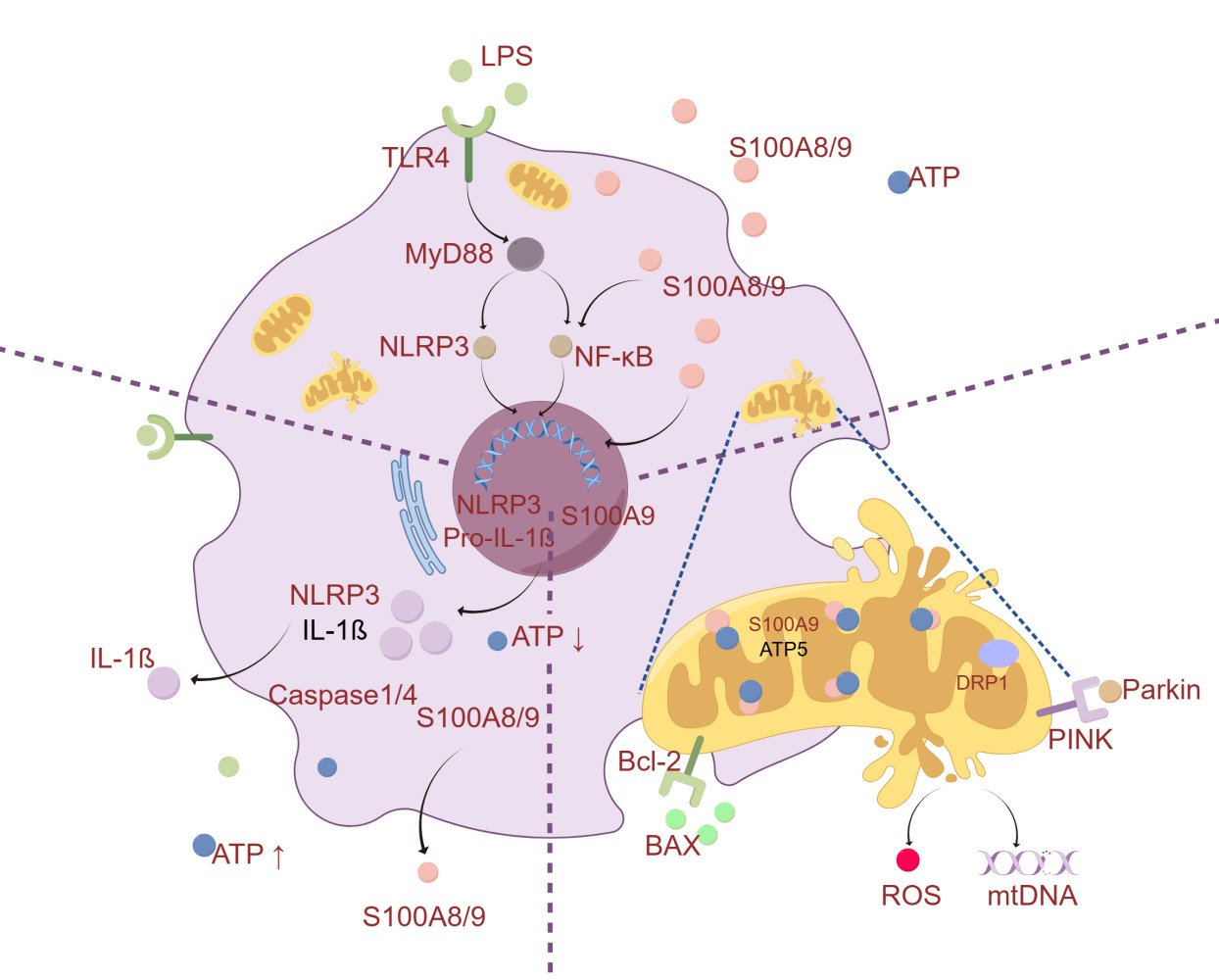
Introduction
Sepsis-induced cardiomyopathy (SICM) is a reversible cardiac dysfunction caused by sepsis and the most prevalent complication, characterized by reduced ejection fraction and poor response to volume resuscitation and vasopressors.1,2 The incidence rate of sepsis cardiomyopathy accounts for approximately 10–70% of sepsis patients,3,4 with a staggering mortality rate of 70–90%.5,6 The pathological mechanism of SICM is highly intricate and encompasses several key factors, including myocardial depression,7,8 mitochondrial dysfunction,9,10 inflammation response,11,12 oxidative stress,13–15 and calcium responsiveness.16,17 Unfortunately, there is still lack of effective treatments or promising targets for myocardial damage in SICM.
S100A8/9, a damage-associated molecular pattern (DAMP), is one of the most sensitive biomarkers in the inflammatory process18 and is closely related to the severity of various inflammatory diseases and multiple organ dysfunction.19 S100A8/9 is highly expressed in various heart diseases, including myocardial infarction,20,21 myocardial hypertrophy,22 heart failure,23,24 and viral myocarditis,25 but its research in septic cardiomyopathy is still insufficient. There is currently limited understanding of the precise mechanisms by which S100A9 induces septic heart dysfunction and myocardial damage.
Previous studies have shown that S100A9 regulates the oxidative and arachidonic acid metabolism of neutrophils,26,27 in addition to mitochondrial energy metabolism, sepsis-associated liver dysfunction, and hepatocellular carcinoma.28,29 S100A9 induces respiratory dysfunction and mitochondrial fission, which ultimately results in cardiac dysfunction. Based on previous research and bioinformatics analyses, we hypothesized that S100A9 induces mitochondrial dysfunction via oxidative stress, consequently influencing myocardial energy metabolism.
Therefore, this study aimed to clarify whether S100A9 is associated with sepsis-induced cardiac injury and dysfunction, and to further elucidate the potential mechanisms underlying S100A9 inducing oxidative stress and mitochondrial dysfunction, as well as whether blocking S100A9 can inhibit oxidative stress and restore mitochondrial function.
Materials and Methods
Bioinformatics Analysis
Transcriptomic data for SICM were obtained from the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/gds/). Differentially expressed genes (DEGs) between SICM and normal samples were identified using the GEO2R online tool (http://www.ncbi.nlm.nih.gov/geo/geo2r) with the following selection criteria: |log2 (fold change)|≥1.5 and P<0.05. TBtools,30 OmicStudio,31 Gene Ontology (GO) term enrichment, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were used to identify the DEGs and their functions. All curated gene sets (www.broadinstitute.org/gsea/index.jsp) and mitochondrial gene sets (MitoCarta3.0 datasets) were used for enrichment analysis using GSEA software. The STRING database (https://string-db.org/) and Molecular Complex Detection (MCODE) of Cytoscape were used to construct a (PPI) network. HDOCK (http://hdock.phys.hust.edu.cn/)32 was used for molecular blind docking and prediction of binding affinity. The molecular binding modes of the proteins were visualized, analyzed, and mapped using PyMOL.33
Machine Learning
Data from 51 patients were obtained from the GSE79962 dataset, and a neural network (NN) model was constructed using the neural net package in R to identify DEGs. To ensure the reliability, tenfold cross-validation was performed. To optimize the model for clinical applications, genetic algorithms (GAs) were applied with the following parameters (pop_size=30, generations=100, crossover_rate=0.9, and mutation_rate=0.01). The fitness of each GA was based on its prediction accuracy, and the roulette-wheel selection method was used as the selection strategy.
Animals and Experimental Procedures
C57BL/6 mice and S100A9 knockout mice were obtained from the Animal Center of Wenzhou Medical University and GemPharmatech (T014553, Nanjing, China). All experimental protocols and methods were approved by the Laboratory Animal Ethics Committee of the Wenzhou Medical University (WYYY-AEC-YS-2022-0381). A mouse model of sepsis-induced cardiomyopathy was established by an intraperitoneal LPS injection (10 mg/kg, Sigma). All the applied procedures followed the Chinese guidelines for the welfare of the laboratory animals (GB/T 35823–2018).
Echocardiographic Assessment
Mice were anesthetized with 2% isoflurane mixed with oxygen during echocardiography. Using a Vevo 2100 ultrasound machine (VisualSonics, Canada) equipped with an MS-550D probe to analyze cardiac function. The mouse heart was imaged in the two-dimensional mode in the parasternal short axis view. The LV dimensions in diastole (LVDd) and systole (LVDs), percentage ejection fraction (EF%) and percentage fractional shortening (FS%) were estimated using M-mode measurements.34
Cell Culture and Treatments
Primary cardiomyocytes were isolated from mice hearts using previously described methods.35 H9C2 cells were purchased from the Shanghai Institute of Biochemistry and Cell Biology, and cultured at a density of 5×104 cells/cm2 in Dulbecco’s modified Eagle’s medium (DMEM, Thermo Fisher Scientific, USA) supplemented with 10% fetal bovine serum (Gibco, USA) and 1% penicillin-streptomycin (Solarbio, China) in a 5% CO2 incubator at 37 °C. After 12 h of starvation, the cardiomyocytes were stimulated with LPS (10 μg/mL) for 12 h to mimic the myocardial injury model. S100A9 was overexpressed or knocked down using lentiviral transfection technology (GenEpharma, China).
Hematoxylin-Eosin (HE) Staining
Heart tissue was fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned into 5-micrometer thick sections. Sections were stained according to the instructions of the HE staining kit (Solarbio, China). Extract 20-fold and 40-fold images in similar myocardium positions, ImageJ software was used to identify the red area (cytoplasmic) and purple area (nuclear) and calculate the cytoplasm-to-nucleus ratio.
Transmission Electron Microscope
The heart tissue was perfused retrogradely and fixed in 2.5% glutaraldehyde. After fixation, samples were washed, dehydrated, embedded, and prepared for ultrathin sectioning. Myocardial mitochondria were observed at the ultrastructural level using a transmission electron microscope (HT7800, Hitachi, Tokyo, Japan) at 80 kV. The morphology of myocardial fibers, mitochondria, and autophagosomes was observed. At least five visual fields per sample were randomly selected to analyze.
Mitochondrial Membrane Potential (MMP)
Cells were incubated with tetramethylrhodamine (TMRE, Beyotime, China) for 20 min following the detection kit instructions. Cells with high MMP levels emitted bright red fluorescence. Flow cytometry (FACSCanto II, BD, USA), a fluorescence microplate reader (ID5, Molecular Devices, USA), and fluorescence microscopy (Olympus, Tokyo, Japan) were used to detect the fluorescence intensity of the TMRE.
ATP and NADP+/NADPH Luciferase Assay
Cardiomyocytes and supernatant were homogenized with ATP lysis buffer, followed by centrifugation at 4 °C. The supernatant was mixed with the ATP detection working solution in a black 96-well plate. ATP levels were measured by fluorescence intensity using a fluorescence microscope per the manufacturer’s instructions (Promega, USA). The NADP+/NADPH content in the cells was assessed using the NADP/NADPH-Glo assay kit per the manufacturer’s protocol (Promega, USA). Fluorescence was measured using a fluorescence microplate reader and the NADP+ and NADPH levels were normalized to those of the control.
Mitochondrial ROS and NO Level
Intracellular levels of ROS and NO were measured using dihydroethidium (DHE, Beyotime, China) and 1,2-Diaminoanthraquinone (DAA AAT Bioquest, USA) staining per the manufacturer’s instructions. Cardiomyocytes were treated with DHE or DAA in the dark for 20 min at 37 °C. Cells with high levels of superoxide anions and nitric oxide emit red fluorescence. Flow cytometry, fluorescence microplate reader, and fluorescence microscopy were used to detect the fluorescence levels.
DNA/RNA Extraction and Library Construction and Sequencing
Genomic DNA was extracted using a TIANamp Genomic DNA kit (DP304, TIANGEN, China) and quantified using a NanoDrop and Qubit. Total RNAs were extracted using a TRIzol reagent and an RNA extraction kit (DP451, TIANGEN, China) following the manufacturer’s instructions. After purification, mRNA was fragmented into small pieces using divalent cations at elevated temperatures. The cleaved RNA fragments were then used to construct a cDNA library using an Illumina RNA ligation-based method (Illumina, USA). Paired-end sequencing (PE150) was performed on a NovaSeq 6000 (LC-Bio Technology CO, China), following the manufacturer’s recommended protocol.
Quantitative Real-Time Polymerase Chain Reaction (qPCR)
Reverse transcription was performed using the ReverseAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, USA) and CFX96 PCR detection system (Bio-Rad, USA). The relative mitochondrial gene copy number (mtDNA to nDNA) and mRNA expression were measured by qPCR using SYBR Green Supermix (Bio-Rad, USA), per the manufacturer’s instructions. GAPDH was used as a reference for mRNA expression quantification and normalized to the control group. The relative expression level was calculated by the 2−ΔΔCT method, as previously described.36 The specific primers were listed in Supplementary Table 1.
Colocalization
Mitochondria were probed with the MitoTracker Red reagent for 30 min before immunofluorescence staining. H9C2 cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton-100, and blocked with 1% BSA, as described in the manufacturer’s protocol. ATP5 and S100A9 were probed with the corresponding mouse antibody (1:200), followed by Alexa Fluor 488-conjugated secondary antibody, and rabbit antibody followed by Fluor 647-conjugated secondary antibody. The nuclei were stained with Hoechst 33342 for 10 min. Colocalization of nuclei (blue), ATP5 (green), and S100A9 (light red) was observed using confocal Microscopy.
Western-Blot Analysis
Protein concentrations in the myocardial tissue or cardiomyocyte lysates were determined using the BCA method. Quantitative proteins were separated by SDS-PAGE and transferred to PVDF membranes, as previously described.35 Supplementary Table 2 shows the specific antibodies. Signals were developed using enhanced chemiluminescence (Millipore) and iBright systems (Thermo Fisher Scientific, USA).
Co-Immunoprecipitation (Co-IP) and Mass Spectrometry (MS)
Co-IP was performed using antibodies against S100A9, S100A8, IgG, and the Immunoprecipitation Kit (Beyotime, China). The antibody working solution (10 μg/mL) was incubated with 20 µL of Protein A+G-coated magnetic beads at 4 °C with gentle shaking overnight. The myocardial tissue protein suspension was mixed and incubated with magnetic beads at room temperature for 2 h with gentle shaking. Immune complex-bound beads were washed three times with PBS. Proteins were eluted with SDS-PAGE loading buffer at 95 °C. The eluted proteins were analyzed by immunoblotting and mass spectrometry with technical support from the Biotech Corporation. An Orbitrap-Fusion mass spectrometer (Thermo Scientific) accompanied by an Easy-nLC 1000 (Thermo Scientific) was used to identify the co-IP proteins. Specific antibodies for Western-blot, IHC and Co-IP were listed in Supplementary Table 2.
Sepsis Patients with Clinical Information
Blood samples and clinical data of 44 patients with sepsis were obtained from the First Affiliated Hospital of Wenzhou Medical University. Our study was approved by the Ethics Committee of the First Affiliated Hospital of Wenzhou Medical University (KY−2023−019) and conformed to the good clinical practice guidelines and the ethical guidelines of the declaration of helsinki. The inclusion and exclusion criteria for sepsis patients were based on the International Consensus on Sepsis and Septic Shock.37 The inclusion criteria of the study were specified as follows: Sepsis patients with SOFA ≥ 2; aged more than 18 years, and patients whose relatives agreed to provide written informed consent. The exclusion criteria were as follows: patients with malignant cancer or end-stage cancer; pregnant or lactating patients; patients with severe heart disease; patients with autoimmune diseases requiring immunosuppressive or hormonal agents; and patients with a poor compliance or serious adverse events.
Statistical Analysis
Statistical analyses and visualization were performed using GraphPad Prism 8.0. One-way analysis of variance (ANOVA) was used to assess significant differences between the means of multiple samples. A t-test was used to calculate the differences between the two groups. The data were presented as Mean±SEM, and P< 0.05 was considered statistically significant.
Results
Differential Expression Genes in SICM by Bioinformatics Analysis
Nine SICM transcriptome datasets38–45 were collected from multiple genera and platforms in the GEO database, as detailed in the Supplementary Table 3. Figure 1 shows a flow chart of the bioinformatics analysis of the transcriptome datasets. A total of common differentially expressed genes (co-DEGs) were identified at the intersection of all mouse transcriptome datasets: Serpina3n, Lcn2, Ms4a6d, Lrg1, Osmr, Socs3, Fcgr2b, S100A9, S100A8, Casp4, Abca8a, and Nfkbiz, as shown and Figure 2A and B. In addition, 36 co-DEGs were identified at the intersection of the four mouse transcriptome datasets, as shown in Figure 2C and Supplementary Table 4. The GO enrichment analysis of 48 co-DEGs was predominantly associated with various biological processes, including immune system processes, innate immune response, inflammatory response, response to the bacterium, lipopolysaccharide, virus, et al (Figure 2D) The KEGG enrichment analysis revealed that the 48 co-DEGs were mainly linked to IL-17 signaling pathway, TNF signaling pathway, NF-kappa B signaling pathway, et al (Figure 2E) To gain insights into potential PPI among the co-DEGs, the MCODE algorithm in Cytoscape was utilized to screen and identify the PPI network, as presented in Figure 2F.
 |
Figure 1 The flow chart of bioinformatics analysis for SICM transcriptome datasets. Abbreviations: SICM, Sepsis-induced cardiomyopathy; GEO, Gene Expression Omnibus database; GEO2R (GEO to R), an interactive web tool to identify genes that are differentially expressed across experimental conditions in a GEO Series; DEGs, differentially expressed genes; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes. |
 |
Figure 2 Differential expression genes in SICM by bioinformatics analysis. (A) Venn diagram of DEGs in 5 mice transcriptome datasets. The 12 co-DEGs in 5 datasets were marked in red. 36 co-DEGs were common differences expressed in 4 datasets. (B) The Volcano plot of 12 co-DEGs with red indicating high expression and blue indicating low expression. (C) The heatmap of 36 co-DEGs expression in 5 datasets. (D) GO enrichment analysis of 48 co-DEGs with enrichment score. (E) KEGG enrichment analysis of the 48 co-DEGs with enrichment score. (F) PPI network of co-DEGs constructed in STRING databases with Cytoscape. 14 genes including S100A8 and S100A9 showed significant protein interactions. The size of the circle represents the significance. |
S100A9 Were Key Co-DEGs in the Development and Progression of SICM
The expression of co-DEGs, especially S100A8 and S100A9, in rat and human heart tissues is shown as a volcano diagram in Figure 3A and B. The deep machine learning algorithm further confirmed the predictive value of co-DEGs for SICM (Figure 3C). GSE79962 contains 11 normal myocardial samples, 20 SICM samples, and 20 non-SICM samples (comprising 11 ischemic cardiomyopathies and 9 dilated cardiomyopathies). According to the NCBI gene database, the 47 co-DEGs were mapped to functionally identical human genes. Classification efficacy testing of co-DEGs using neural networks (NN) demonstrated strong classification performance for cardiomyopathy (Figure 3D). Notably, 6 genes with the highest weights, namely, DHX58, GADD45G, OSMR, HAMP, SERPINA3N, and S100A9, were identified as significant biomarkers for distinguishing different types of cardiomyopathies (Figure 3E). Subsequently, a genetic algorithm (GA) was introduced to optimize the neural networks and develop a four-gene classification prediction model. After iterations with the prediction accuracy for each progeny, MS4A6, S100A9, ABCA8A, and ANGPTL4 were identified as biomarkers that effectively differentiated the normal myocardium, SICM, and non-SIMC (Figure 3F). In addition, ABCA8A, SERPINA3, NFKBIZ, MS4A6, S100A9, and S100A8 were significantly correlated with cardiac function, including left ventricular ejection fraction (LVEF), left ventricular end-systolic diameter (LVESD), and left ventricular end-diastolic diameter (LVEDD) (Figure 3G).
 |
Figure 3 S100A8/9 were key DEGs in the development and progression of SICM. (A) Volcano plots show the expression of 48 co-DEGs in human and rat sepsis myocardial datasets, with red representing high expression and blue representing low expression. (B) Transcription expression of S100A8 and S100A9 mRNA in human and rat myocardial datasets. (C) Flowchart of machine learning and model detection. (D) Framework of artificial neural networks model for predicting SICM and no-SICM. The input layer contained 47 neurons transformed by NCBI. The output layer had three neurons representing the probability of SICM, normal group, and no-SICM. (E) Classification efficacy testing of co-DEGs using neural networks. DHX58, GADD45G, OSMR, HAMP, SERPINA3N, and S100A9 had the highest selection weight. (F) Diagram of confusion matrix for prediction accuracy of four-gene class prediction model. (G) Correlation analysis between DEGs expression and cardiac function in the GSE79962 dataset. The mRNA transcript levels of ABCA8, SERPIN3, NFKBIZ, MS4A6, S100A8 and S100A9 were closely related to LVEF%, LVESD and LVEDD. *P<0.05. ** P<0.01, ***P<0.001. |
S100A9 Was Associated with Cardiac Dysfunction in Septic Patients
To investigate the correlation between S100A9 expression and cardiac function in patients, our study reviewed 44 patients diagnosed with sepsis after admission to the EICU. The basic characteristics and laboratory tests of sepsis patients were shown in Supplementary Table 5. Patients with septic shock displayed significantly higher S100A9 expression compared to non-shock patients (3.45±2.04 vs 1.97±1.25, P=0.02). High S100A9 expression was associated with an inflammatory response and poorer cardiac function in patients with sepsis, including PCT, CRP, LVEF, pro-BNP, cTNI, and CK-MB, as detailed in Table 1.
 |
Table 1 Correlation of S100A9 with Clinical Characteristics of Sepsis Patients |
S100A9 Knockout Alleviated Myocardial Damage and Cardiac Dysfunction
Detection of inflammatory marker and S100A8/9 expression to demonstrate successful simulation of sepsis-induced cardiomyopathy, as detailed in the Supplementary Tables 1 & 2. Nucleic acid gel electrophoresis confirmed the absence of the S100A9 band (413 bp) in the knockout allele, while observed in the wild-type allele (Figure 4A). Compared to the wild-type control and LPS groups, the protein expression of S100A9 was significantly reduced in the S100A9KO group (Figure 4B). In addition, the mRNA transcription of inflammation and heart failure markers in the myocardium, such as IL1, IL6, TNFa, ANP, BNP, and MYHC, was considerably reduced in the S100A9KO group compared with that in the wild-type sepsis group (Figure 4C). Meanwhile, sepsis-induced cardiomyocyte swelling, especially the proportions of the cytoplasm and nucleus, were correspondingly alleviated in the S100A9KO group, as shown in Figure 4D-F. Transthoracic echocardiography indicated that the decrease in EF% and FS% induced by sepsis was partially relieved by the S100A9 knockout, with statistically significant differences, as illustrated in Figure 4E-H. S100A9 knockout alleviates sepsis-induced cardiomyopathy and reverses myocardial injury.
 |
Figure 4 Knocking out the S100A9 gene alleviated myocardial damage and cardiac dysfunction. (A) Identification of the S100A9 gene by PCR. Position of the S100A9 gene (413 bp) on agarose gel electrophoresis. (B) The protein expression of S100A9 and GAPDH in myocardium. (C) The mRNA transcription level of IL1, IL6, TNFα, ANP, BNP, and MYH transcription in primary cardiomyocytes. (D) HE staining for myocardial damage in SICM. Extract 20-fold and 40-fold images in similar myocardium positions, use imagJ software to identify the red area (cytoplasmic) and purple area (nuclear), and calculate the ratio of cytoplasm to the nucleus. (E) Transthoracic echocardiography for myocardial dysfunction in SICM. The left ventricle was imaged using the cross-sectional mode with parasternal long/short-axis views. (F) Statistical histogram of the ratio of cytoplasm to nucleus. (G) Serum CKMB content at standard protein concentration in SICM. (H) Histograms of cardiac function with EF% and FS%. The data were shown as the Mean ± SEM, from n=6 per group. Abbreviations: IL1, interleukin 1; IL6, interleukin 6; TNFa, tumor necrosis factor alpha; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; WT, wild type mice; S100A9KO, S100A9 gene knock out mice; Con, control; LPS, lipopolysaccharides; ANP, natriuretic peptide type A; BNP, natriuretic peptide type B; MYHC, myosin heavy chain; *P<0.05. ** P<0.01, ***P<0.001. |
S100A9 Knockout Alleviated Mitochondrial Damage and Oxidative Stress
Mitochondrial damage and oxidative stress are closely associated with cardiomyopathy exacerbation. The mitochondria of the myocardium in the wild-type control group were well-proportioned, balanced in size, and regularly arranged. The mitochondrial cristae density was determined using transmission electron microscopy. However, the quantity and size heterogeneity of the mitochondria significantly increased in the sepsis group, with swelling and blurring of the mitochondrial cristae and visible medullary structures in individual mitochondria. Compared to the wild-type sepsis group, mitochondrial cristae swelling and hypertrophy were partially alleviated in the S100A9KO group. S100A9 knockout partially alleviated mitochondrial damage and restored the normal structure (Figure 5A). The intensities of DHE and DAA fluorescence, as well as the malondialdehyde (MDA) concentration, increased substantially in the sepsis group, indicating a marked increase in oxidative stress in the myocardium. Conversely, the superoxide dismutase (SOD) concentration in the myocardium was markedly reduced in the sepsis group, which was reversed by S100A9 knockout (Figure 5B-E).
 |
Figure 5 S100A9 knockout alleviated mitochondrial damage and oxidative stress. (A) Transmission electron microscopy detection of mitochondrial morphology in similar myocardium positions. (B and C) Oxidative stress of myocardium. Intracellular ROS and NO were detected by DHE and DAA probes unDer Standard protein concentrations. (D and E) Content of SOD and MDA in myocardial tissue. MDA and SOD at standard protein concentrations were detected by absorbance. *P<0.05. ** P<0.01, ***P<0.001. Abbreviations: DHE, dihydroethidium, peroxide red fluorescent indicator; DAA, 1.2-diaminoanthraquinone, sensitivity, specificity and nontoxic nitric oxide fluorescent probe; ROS, reactive oxygen species; NO, nitric oxide; SOD, superoxide dismutase; MDA, malondialdehyde. |
Effects of S100A9 Knockdown in Primary Myocardial Cells Through Transcriptomics
Wild-type and CRISPR/Cas9 S100A9KO mice were used to isolate primary cardiomyocytes for subsequent bioinformatic analysis, as illustrated in Figure 6A. The quality and purity of the extracted primary myocardial cells were evaluated by fluorescent staining for Actn2 in Supplementary Figure. Compared with the corresponding control group, 2946 genes were significantly affected by LPS, with 1370 genes upregulated and 1576 genes downregulated. Similarly, 1406 genes were affected by the S100A9 knockout, with 595 upregulated and 811 downregulated genes (Figure 6B). There were 238 co-DEGs among the three groups, as shown in Figure 6C. The top 100 DEGs affected by S100A9 knockout were revealed in the heatmap, including the heart damage markers Nppa and Tnni1 (Figure 6D and E).
 |
Figure 6 Effects of S100A9 knockdown on transcriptomics in primary myocardial cells. (A) Flowchart of primary cardiomyocytes extraction and bioinformatics analysis. (B and C) The amount of DEGs between WT-con group, WT-lps group and S100A9KO-lps group, and discriminated up-regulated genes and down-regulated genes. Significantly different genes were screened through P-values<0.05 and |logFC|>1. (D) The top 100 DEGs between WT-lps group and S100A9KO-lps group by Heatmap with red representing high expression and blue representing low expression. (E) The top 20 DEGs between WT-lps group and S100A9KO-lps group by Volcano plot. (F and G) The top 20 of biological process and molecular function by GO enrichment analysis. (H and I) GSEA enrichment analysis of DEGs between WT-lps group and S100A9KO-lps group, including pathways related to myocardial contraction, oxidative stress and mitochondrial function. Abbreviations: WT, wild type mice; S100A9KO, S100A9 gene knock-out mice; Con, Control; LPS, Lipopolysaccharides. NES: normalized enrichment score. |
GO enrichment analysis indicated that the S100A9 knockout significantly affected various biological processes and molecular functions, including the inflammatory response, immune response, NADPH oxidase activator activity, and chemokine activity, as shown in Figure 6F and G. Additionally, KEGG enrichment analysis indicated that the NFKB signaling pathway, Toll-like receptor signaling pathway, Cytokine-cytokine receptor interaction pathway, and the pathways related to cardiac muscle contraction or cardiomyopathy were affected by S100A9 knockout. GSEA revealed that myocardial contractility, oxidative stress, and mitochondrial function were significantly affected by the S100A9 knockout, including muscle contraction, superoxide metabolic process, NADPH oxidase activity, TCA cycle, and respiratory electron transport, as shown in Figure 6H and I and Supplementary Figure.
Effects of S100A9 on H9C2 Cells Viability and Oxidative Stress
S100A9 overexpression and knockdown in H9C2 cells were achieved by lentivirus transfection, with technical support from GenePharma Corporation. S100A9 protein expression in cardiomyocytes significantly increased in the S100A9 overexpression (S100A9OV) group and decreased in the S100A9 knockdown (S100A9KD) group (Figure 7A). The viability of LPS-stimulated cardiomyocytes initially increased and then decreased. Cardiomyocytes in the S100A9KD group demonstrated greater resistance to LPS stimulation (Figure 7B). S100A9 knockdown alleviated LPS-induced myocardial injury and reversed the myocardial injury markers ANP, BNP, and MYHC.(Figure 7C) The oxidative stress in cardiomyocytes was detected using DHE and DAA probes and reached its peak after 12 h of LPS stimulation (Figure 7D and Supplementary Figure). Intracellular oxidative stress levels, detected by flow cytometry, increased in the S100A9OV group after LPS stimulation for 12 h and were reversed by S100A9 knockdown (Figure 7E, F and H).
 |
Figure 7 S100A9 affected H9C2 cells viability and oxidative stress. (A) S100A9 and GAPDH protein levels in the different groups. The histograms in each panel normalized to its corresponding GAPDH. (B) CCK8 assay for detecting cardiomyocyte viability after LPS 3, 6, and 12 hours. (C) Detection of ANP, BNP and MYHC mRNA transcription levels after LPS 12h; (D) Detection of oxidative stress induced by LPS through DHE probe within 0–48 hours. (E and F) Detection of DHE fluorescent probes intensity by flow cytometry. (G) NADP+/NADPH-Glo reagent kit detected NADP+/NADPH content in cardiomyocytes, and relative quantification was performed with the control group based on fluorescence intensity. (H) Immunofluorescence of DHE probes in H9C2 cells after LPS stimulation 12 hours. (I) Protein levels of NLRP3, phosphorylated and total of NFkb, Caspase 1, and bACT were determined by Western blotting using specific antibodies. *P<0.05. ** P<0.01, ***P<0.001. Abbreviations: Con, Control; LPS, Lipopolysaccharides, NC, negative control, empty Lentiviral vectors by overexpressing or knocking down the S100A9 gene. OV, Overexpression of S100A9 gene; KD, Knockdown of S100A9 gene; ROS, reactive oxygen species; NADPH, Nicotinamide adenine dinucleotide phosphate; NLRP3, NLR family, pyrin domain containing 3; NFKB, nuclear factor kappa B, p-NFKB, Phosphorylated NFKB; bACT, actin beta. |
Bioinformatics analysis demonstrated that S100A9 activates NADPH oxidase (NOX) to induce oxidative stress in myocardial cells. S100A9 knockdown inhibited NOX activation and reversed the increase in the NADP+/NADPH ratio, as shown in Figure 7G. As shown in Figure 7I, the expression of NLRP3, p-NFKB, and Caspase1 proteins was significantly increased in the S100A9OV group compared to the S100A9NC group, while it was decreased in the S100A9KD group.
Effects of S100A9 on Mitochondrial Function and Energy Metabolism
The mitochondrial membrane potential (MMP) was detected by the TMRE probe and reached its minimum at approximately 12–24 h, as detailed in the Supplementary Figure. Fluorescence imaging indicated that S100A9 overexpression resulted in a significant reduction in membrane potential (red) at LPS 12h, as shown in Figure 8A. S100A9 knockout effectively alleviated the decrease in MMP and partially reversed the reduced mitochondrial function, as determined by flow cytometry. (Figure 8B and C) Intracellular and extracellular ATP levels were measured to assess mitochondrial energy metabolism. Abnormal energy metabolism induced by LPS is mainly manifested by a decrease in intracellular ATP content and an increase in ATP leakage. S100A9 overexpression exacerbates intracellular energy metabolism dysfunction and mitochondrial ATP leakage (Figure 8D and E).
 |
Figure 8 S100A9 induces mitochondrial damage and energy metabolism impairment. (A) MMP fluorescence staining by TMRE probe (red) and Hoechst 33342 (blue). (B and C) Flow cytometry detection of TMRE fluorescence intensity in H9C2 cells. (D and E) Detection of intracellular and extracellular ATP content by fluorescence luminescence method. (F-H) Mitochondrial mRNA transcript levels of TFAM, TFBM, ATP6, ATP8, COX1, COX2 and COX3 by qPCR. The above statistical results were compared with the corresponding control group. *P<0.05. ** P<0.01, ***P<0.001. Abbreviations: Con, Control; LPS, Lipopolysaccharides; NC, negative control; OV, Overexpression of S100A9 gene; KD, Knockdown of S100A9 gene; TMRE, tetramethylrhodamine, specifically recognize mitochondrial membrane potentials; MMP, mitochondrial membrane potential. |
The mitochondrion is a semiautonomous organelle responsible for protein synthesis and energy metabolism and relies heavily on mtDNA gene copies and proper transcription. The mtDNA copy number was significantly increased in sepsis cardiomyopathy, as detailed in the Supplementary Figure, whereas the transcription levels of mitochondrial proteins decreased in the S100A9OV-LPS group, including mitochondrial transcription factors (TFAM and TFBM), mitochondrial ATP synthases (ATP6 and ATP8), and cytochrome C oxidase factors (COX1, COX2, and COX3) (Figure 8F-H).
S100A9-Induced Oxidative Stress and Mitochondrial Damage Alleviated by Paquinimod
Paquinimod, a specific S100A8/9 inhibitor, had a minor impact on cardiomyocyte viability at concentrations below 100 uM, as shown in Figure 9A. Paquinimod effectively reduced oxidative stress in S100A9 overexpression H9C2 cells with preconditioned concentrations ranging from 12.5 uM to 100 Um, and did not attenuate NO activity induced by LPS. (Figure 9B and C) The fluorescence image of the DHE probe in cardiomyocytes with paquinimod pretreatment (50–100 uM) was presented in Figure 9D. Compared to the S100A9NC group, the expression of Drp1, Pink1, and Parkin proteins was dramatically enhanced in the S100A9OV group and reduced in the S100A9KD group, as shown in Figure 9E and F. Moreover, the application of paquinimod pretreatment (50–100 uM) can significantly inhibit the corresponding mitochondrial division and autophagy proteins in S100A9 overexpression cardiomyocytes (Figure 9G and H).
 |
Figure 9 S100A9-induced oxidative stress and mitochondrial damage alleviated by Paquinimod. (A) Detection cardiomyocyte viability in different concentrations of paquinimod at 12 hours. (B and C) The effect of paquinimod pretreatment 2 hours with different concentrations on oxidative stress levels in cardiomyocytes. (D) Paquinimod pretreatment on DHE staining in S100A9 overexpression cardiomyocytes. (E and F) Protein levels of Drp1, Pink1, Parkin, S100A9, and GAPDH were determined by Western blotting. The above statistical results were compared with the corresponding control group. *P<0.05. ** P<0.01, ***P<0.001. Abbreviations: Con, Control; LPS, Lipopolysaccharides; NC, negative control; OV, Overexpression of S100A9 gene; KD, Knockdown of S100A9 gene; PAQ, paquinimod, a specific inhibitor of S100A8/9; ROS, reactive oxygen species; NO, nitric oxide. |
Detection of Proteins Interacting with S100A8/9
Co-immunoprecipitation (Co-IP) of S100A9 and S100A8 was used to reveal the interactions between myocardial proteins. After excluding potential interference from immunoglobulins and keratin, 45 proteins interacting with S100A8/9 were identified using mass spectrometry (MS). (Figure 10A). These proteins were mainly enriched in processes related to mitochondria, oxidoreductase activity (NAD/NADP+), muscle contraction, cellular respiration, ATP metabolism, and mitochondrial electron transport (Figure 10B). KEGG enrichment analysis revealed that these proteins are involved in various types of pathological cardiomyopathy, metabolism, and reactive oxygen species pathways. (Figure 10C) Particularly, ACTA2 (actin, smooth muscle) is involved in vascular contractility, myocardial contraction, and blood pressure homeostasis. Mitochondrial ATP5 (ATP5F1A and ATP5F1B) was also related to energy metabolism and mitochondrial membrane potential, as shown in Table 2 and 3. Based on the docking scores provided by HDOCK, we selected the best-predicted binding mode to evaluate the interaction between ATP5 and S100A8/9, as shown in Figure 10D, E and Supplementary Tables 6-8. Immunofluorescence demonstrated co-localization of S100A9 and ATP5 proteins in cardiomyocytes. (Figure 10F) We validated the interaction between S100A9 and ATP5 proteins using co-immunoprecipitation (Figure 10G).
 |
Table 2 Identification by MS: the Top Five Proteins coIP with S100A8 Protein |
 |
Table 3 Identification by MS: the Top Five Proteins coIP with S100A9 Protein |
 |
Figure 10 Detection of proteins interacting with S100A8/9. (A) Identification of proteins interacting with S100A8/9 in myocardial by co-IP and MS. (B) GO enrichment analysis of interacting proteins with gene count. (C) KEGG enrichment analysis of interacting proteins with gene count. (D and E) Molecular docking between S100A8/9 and ATP5 were visualized by PyMOL. (F) Co-localization of S100A9 (shown in light red) and ATP5 (green) proteins in cardiomyocytes. (G) Co-IP results of S100A9 and ATP5 proteins in H9C2 cells. The above statistical results were compared with the corresponding control group. Abbreviations: Co-IP, co-immunoprecipitation; MS, mass spectrometry, ATP5F1A, ATP synthase F1 subunit alpha; ATP5F1B, ATP Synthase F1 Subunit Beta. |
Discussion
SICM is a life-threatening condition that is commonly observed in critically ill patients. Unfortunately, early identification and effective treatments for this condition are currently lacking, leading to high mortality rates. To address this gap, we utilized transcriptome data from SICM models across multiple genera and platforms to conduct a bioinformatics analysis. Twelve co-DEGs were identified as key pathogenic genes in sepsis cardiomyopathy, including Serpina3n, Lcn2, Ms4a6d, Lrg1, Osmr, Socs3, Fcgr2b, S100a9, S100a8, Casp4, Abca8a, and Nfkbiz. Bioinformatic analysis and machine learning classification prediction models have consistently highlighted the crucial role of S100A8/9 in the diagnosis and prognosis of SICM.
S100A8/9 is not only a biomarker of SICM development but is also closely related to the clinical prognosis of sepsis patients.46–49 In our study, we found a clear correlation between elevated S100A9 levels and inflammatory responses and poorer cardiac function in patients with sepsis. Similarly, S100A8/9 levels were significantly increased in patients with acute myocardial infarction, and associated with the occurrence of major adverse cardiovascular events.20 The above high-throughput transcriptional data and clinical evidence support our comprehensive investigation into the role of S100A9 in cardiomyopathy development.
S100A8/9 proteins are produced by circulating immune cells and myocardial cells and act as damage-associated molecular patterns (DAMPs) that trigger inflammatory responses50,51 and myocardial damage.52,53 The knockout of S100A9 resulted in the disruption of the coordination structure of the heterodimer, ultimately leading to the degradation of S100A8/9 functionality.54 Previous studies have predominantly focused on the release of S100A8/9 by immune cells in response to inflammatory cascade reactions.55 Previous studies found that multiple inflammatory biomarkers were involved in the occurrence and development of cardiovascular and cerebrovascular diseases.56,57 The activation of TLR4 or RAGE signaling pathways induced by inflammatory biomarkers S100A8/9 exacerbate myocardial damage.53,58,59 Indeed, the inflammatory damage induced by S100A8/9 may not be the sole factor contributing to heart dysfunction in SICM.58
Transcriptome analysis of primary cardiomyocytes revealed that S100A9 exacerbated myocardial damage by inducing mitochondrial dysfunction and NOX-mediated oxidative stress. Mitochondria account for approximately 30% of the cell content and provide over 95% of the ATP energy required in cardiomyocytes.60 Mitochondrial dysfunction plays a critical role in heart failure and various types of cardiomyopathy.61–66 Mitochondria are semi-autonomous organelles in eukaryotic cells that constantly undergo fusion and fission events in response to energy demand.67,68 Mitochondrial function relies on relatively independent mtDNA replication and mRNA transcription processes.69–71 The number of mitochondria and mtDNA gene copies was significantly increased in sepsis-induced cardiomyopathy. Interestingly, the transcription of mitochondrial transcription factors and ATP synthase was decreased in cardiomyocytes overexpressing S100A9. An increase in the mtDNA gene copy number and a decrease in mitochondrial mRNA transcription may be direct mitochondrial dysfunction indicators. Our research demonstrated that S100A9 overexpression leads to more severe mitochondrial damage, resulting in reduced MMP and ATP synthesis, disrupted mtDNA replication and transcription, and increased mitochondrial division and autophagy. Accompanied by the opening of the mitochondrial permeability transition pore, the extravasation of mitochondrial contents, such as ATP and metabolites, aggravates pyroptosis, inflammatory damage, and oxidative stress in sepsis-induced cardiomyopathy.72–74
Oxidative stress induced by sepsis originates from reactive oxygen species in the mitochondria, NADPH oxidase (NOX), and nitric oxide synthase (NOS) in cardiomyocytes.75–77 S100A9 deletion dramatically reduces the transcription level of NOX in cardiomyocytes. S100A9 knockdown alleviates NOX-induced oxidative stress in H9C2 cells. Furthermore, it is important to note that NOX also consumes intracellular charge balance and impairs mitochondrial electron chain transmission, further exacerbating mitochondrial energy supply disorders. It is important to note that NOX also depletes the intracellular charge and hinders the mitochondrial respiratory chain, further exacerbating mitochondrial energy supply disorders.76,78,79 Activation of NOX by S100A9 leads to electron leakage and aggravation of mitochondrial oxidative stress. Inhibition of NOX prevents sepsis-induced myocardial damage by improving calcium handling and mitochondrial function.80 These studies indirectly support the validity and importance of our research.
As demonstrated by co-IP and MS, ACTA2 and ATP5 proteins were the first discovered S100A8/9 proteins targeting the myocardium and mitochondria. Moreover, these Results provide further evidence for the close association between S100A8/9 and various critical processes in SICM, including oxidative stress, cardiac contractile function, and mitochondrial energy metabolism. S100A9-induced inflammatory damage, oxidative stress, and mitochondrial dysfunction result in complex interactions and a vicious cycle, ultimately exacerbating sepsis-induced myocardial damage. The interplay between these three elements is mutually causal and they affect each other, thereby exacerbating the SICM pathophysiology. However, our research demonstrated that paquinimod, a specific S100A8/9 inhibitor, can partially disrupt this vicious cycle and alleviate mitochondrial damage and oxidative stress in septic cardiomyocytes.
Limitations
This study supplemented the relevant content of transcriptome sequencing of primary cardiomyocytes. Unfortunately, the mechanism of S100A9-induced mitochondrial damage and oxidative stress was validated using the H9C2 cells rather than primary cardiomyocytes. In addition, retrospective clinical studies included relatively few patients with sepsis, and larger prospective clinical cohort studies are needed.
Conclusion
The interaction of S100A9 with ATP5 exacerbates myocardial damage in sepsis by inducing mitochondrial dysfunction and oxidative stress.
Data Sharing Statement
The datasets generated and/or analyzed in the current study are available from the corresponding author upon reasonable request.
Funding
This work was supported by Scientific Research Fund of Zhejiang Provincial Education Department (Y202250204), the Key R&D Program Projects of Zhejiang Province (2021C03072), the National Natural Science Foundation of China (82272201 and 82272202), Zhejiang Province Clinical Key Specialized Emergency Medicine Department, and the key specialty of traditional Chinese Medicine of Zhejiang Provincial in the 13th Five-Year Plan period (Emergency Department).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hollenberg SM, Singer M. Pathophysiology of sepsis-induced cardiomyopathy. Nat Rev Cardiol. 2021;18:424–434. doi:10.1038/s41569-020-00492-2
2. Liu YC, Yu MM, Shou ST, et al. Sepsis-Induced Cardiomyopathy: mechanisms and Treatments. Front Immunol. 2017;8:1021. doi:10.3389/fimmu.2017.01021
3. Vieillard-Baron A, Caille V, Charron C, et al. Actual incidence of global left ventricular hypokinesia in adult septic shock. Crit Care Med. 2008;36:1701–1706. doi:10.1097/CCM.0b013e318174db05
4. Beesley SJ, Weber G, Sarge T, et al. Septic Cardiomyopathy. Crit Care Med. 2018;46:625–634. doi:10.1097/CCM.0000000000002851
5. Rhodes A, Evans LE, Alhazzani W, et al. Surviving Sepsis Campaign: international Guidelines for Management of Sepsis and Septic Shock: 2016. Intensive Care Med. 2017;43:304–377. doi:10.1007/s00134-017-4683-6
6. Ravikumar N, Sayed MA, Poonsuph CJ, et al. Septic Cardiomyopathy: from Basics to Management Choices. Curr Probl Cardiol. 2021;46:100767. doi:10.1016/j.cpcardiol.2020.100767
7. Court O, Kumar A, Parrillo JE, et al. Clinical review: myocardial depression in sepsis and septic shock. Crit Care. 2002;6:500–508. doi:10.1186/cc1822
8. Shen XD, Zhang HS, Zhang R, et al. Progress in the Clinical Assessment and Treatment of Myocardial Depression in Critically Ill Patient with Sepsis. J Inflamm Res. 2022;15:5483–5490. doi:10.2147/JIR.S379905
9. Stanzani G, Duchen MR, Singer M. The role of mitochondria in sepsis-induced cardiomyopathy. Biochim Biophys Acta Mol Basis Dis. 2019;1865:759–773. doi:10.1016/j.bbadis.2018.10.011
10. Lin Y, Xu Y, Zhang Z. Sepsis-Induced Myocardial Dysfunction (SIMD): the Pathophysiological Mechanisms and Therapeutic Strategies Targeting Mitochondria. Inflammation. 2020;43:1184–1200. doi:10.1007/s10753-020-01233-w
11. Cai ZL, Shen B, Yuan Y, et al. The effect of HMGA1 in LPS-induced Myocardial Inflammation. Int J Biol Sci. 2020;16:1798–1810. doi:10.7150/ijbs.39947
12. Abdullah M, Berthiaume JM, Willis MS. Tumor necrosis factor receptor-associated factor 6 as a nuclear factor kappa B-modulating therapeutic target in cardiovascular diseases: at the heart of it all. Transl Res. 2018;195:48–61. doi:10.1016/j.trsl.2017.10.012
13. Galley HF. Oxidative stress and mitochondrial dysfunction in sepsis. Br J Anaesth. 2011;107:57–64. doi:10.1093/bja/aer093
14. Wang R, Xu Y, Fang Y, et al. Pathogenetic mechanisms of septic cardiomyopathy. J Cell Physiol. 2022;237:49–58. doi:10.1002/jcp.30527
15. Pei H, Qu J, Chen JM, et al. The effects of antioxidant supplementation on short-term mortality in sepsis patients. Heliyon. 2024;10:e29156. doi:10.1016/j.heliyon.2024.e29156
16. Pinto BB, Dyson A, Umbrello M, et al. Improved Survival in a Long-Term Rat Model of Sepsis Is Associated With Reduced Mitochondrial Calcium Uptake Despite Increased Energetic Demand. Crit Care Med. 2017;45:e840–e848. doi:10.1097/CCM.0000000000002448
17. Zhang C, Mo M, Ding W, et al. High-mobility group box 1 (HMGB1) impaired cardiac excitation-contraction coupling by enhancing the sarcoplasmic reticulum (SR) Ca(2+) leak through TLR4-ROS signaling in cardiomyocytes. J Mol Cell Cardiol. 2014;74:260–273. doi:10.1016/j.yjmcc.2014.06.003
18. Shankar M, Uwamahoro N, Backman E, et al. Immune Resolution Dilemma: host Antimicrobial Factor S100A8/A9 Modulates Inflammatory Collateral Tissue Damage During Disseminated Fungal Peritonitis. Front Immunol. 2021;12:553911. doi:10.3389/fimmu.2021.553911
19. Pirr S, Dauter L, Vogl T, et al. S100A8/A9 is the first predictive marker for neonatal sepsis. Clin Transl Med. 2021;11:e338. doi:10.1002/ctm2.338
20. Li Y, Chen B, Yang X, et al. S100a8/a9 Signaling Causes Mitochondrial Dysfunction and Cardiomyocyte Death in Response to Ischemic/Reperfusion Injury. Circulation. 2019;140:751–764. doi:10.1161/CIRCULATIONAHA.118.039262
21. Pei XM, Tam BT, Sin TK, et al. S100A8 and S100A9 Are Associated with Doxorubicin-Induced Cardiotoxicity in the Heart of Diabetic Mice. Front Physiol. 2016;7:334. doi:10.3389/fphys.2016.00334
22. Wei X, Wu B, Zhao J, et al. Myocardial Hypertrophic Preconditioning Attenuates Cardiomyocyte Hypertrophy and Slows Progression to Heart Failure Through Upregulation of S100A8/A9. Circulation. 2015;131:1506–1517. doi:10.1161/CIRCULATIONAHA.114.013789
23. Marinkovic G, Grauen Larsen H, Yndigegn T, et al. Inhibition of pro-inflammatory myeloid cell responses by short-term S100A9 blockade improves cardiac function after myocardial infarction. Eur Heart J. 2019;40:2713–2723. doi:10.1093/eurheartj/ehz461
24. Feng L, Li G, An J, et al. Exercise Training Protects Against Heart Failure Via Expansion of Myeloid-Derived Suppressor Cells Through Regulating IL-10/STAT3/S100A9 Pathway. Circ Heart Fail. 2022;15:e008550. doi:10.1161/CIRCHEARTFAILURE.121.008550
25. Muller I, Vogl T, Pappritz K, et al. Pathogenic Role of the Damage-Associated Molecular Patterns S100A8 and S100A9 in Coxsackievirus B3-Induced Myocarditis. Circ Heart Fail. 2017:10. doi:10.1161/CIRCHEARTFAILURE.117.004125
26. Sroussi HY, Lu Y, Villines D, et al. The down regulation of neutrophil oxidative metabolism by S100A8 and S100A9: implication of the protease-activated receptor-2. Mol Immunol. 2012;50:42–48. doi:10.1016/j.molimm.2011.12.001
27. Kerkhoff C, Klempt M, Kaever V, et al. The two calcium-binding proteins, S100A8 and S100A9, are involved in the metabolism of arachidonic acid in human neutrophils. J Biol Chem. 1999;274:32672–32679. doi:10.1074/jbc.274.46.32672
28. Zhang Y, Wu F, Teng F, et al. Deficiency of S100A9 Alleviates Sepsis-Induced Acute Liver Injury through Regulating AKT-AMPK-Dependent Mitochondrial Energy Metabolism. Int J Mol Sci. 2023;24. doi:10.3390/ijms24032112
29. Zhong C, Niu Y, Liu W, et al. S100A9 Derived from Chemoembolization-Induced Hypoxia Governs Mitochondrial Function in Hepatocellular Carcinoma Progression. Adv Sci (Weinh). 2022;9:e2202206. doi:10.1002/advs.202202206
30. Chen C, Chen H, Zhang Y, et al. TBtools: an Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol Plant. 2020;13:1194–1202. doi:10.1016/j.molp.2020.06.009
31. Lyu F, Han F, Ge C, et al. OmicStudio: a composable bioinformatics cloud platform with real‐time feedback that can generate high-quality graphs for publication. iMeta. 2023.
32. Yan Y, Tao H, He J, et al. The HDOCK server for integrated protein-protein docking. Nat Protoc. 2020;15:1829–1852. doi:10.1038/s41596-020-0312-x
33. Rosignoli S, Paiardini A. Boosting the Full Potential of PyMOL with Structural Biology Plugins. Biomolecules. 2022;12. doi:10.3390/biom12121764
34. Di Lascio N, Kusmic C, Stea F, et al. Longitudinal micro-ultrasound assessment of the ob/ob mouse model: evaluation of cardiovascular, renal and hepatic parameters. Int J Obes Lond. 2018;42:518–524. doi:10.1038/ijo.2017.219
35. Pei H, Wang W, Zhao D, et al. G Protein-Coupled Estrogen Receptor 1 Inhibits Angiotensin II-Induced Cardiomyocyte Hypertrophy via the Regulation of PI3K-Akt-mTOR Signalling and Autophagy. Int J Biol Sci. 2019;15:81–92. doi:10.7150/ijbs.28304
36. Pei H, Chen J, Qu J, et al. S100A9 exacerbates sepsis-induced acute lung injury via the IL17-NFkappaB-caspase-3 signaling pathway. Biochem Biophys Res Commun. 2024;710:149832. doi:10.1016/j.bbrc.2024.149832
37. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315:801–810. doi:10.1001/jama.2016.0287
38. Ashton KJ, Reichelt ME, Mustafa SJ, et al. Transcriptomic effects of adenosine 2A receptor deletion in healthy and endotoxemic murine myocardium. Purinergic Sig. 2017;13:27–49. doi:10.1007/s11302-016-9536-1
39. Ektesabi AM, Mori K, Tsoporis JN, et al. Mesenchymal Stem/Stromal Cells Increase Cardiac miR-187-3p Expression in a Polymicrobial Animal Model of Sepsis. Shock. 2021;56:133–141. doi:10.1097/SHK.0000000000001701
40. Li CC, Hsiang CY, Lo HY, et al. Genipin inhibits lipopolysaccharide-induced acute systemic inflammation in mice as evidenced by nuclear factor-kappaB bioluminescent imaging-guided transcriptomic analysis. Food Chem Toxicol. 2012;50:2978–2986. doi:10.1016/j.fct.2012.05.054
41. Dos Santos CC, Gattas DJ, Tsoporis JN, et al. Sepsis-induced myocardial depression is associated with transcriptional changes in energy metabolism and contractile related genes: a physiological and gene expression-based approach. Crit Care Med. 2010;38:894–902. doi:10.1097/CCM.0b013e3181ce4e50
42. Han YC, Xie HZ, Lu B, et al. Lipopolysaccharide Alters the m6A Epitranscriptomic Tagging of RNAs in Cardiac Tissue. Front Mol Biosci. 2021;8:670160. doi:10.3389/fmolb.2021.670160
43. Tran TT, Mathieu C, Torres M, et al. Effect of landiolol on sex-related transcriptomic changes in the myocardium during sepsis. Intensive Care Med Exp. 2019;7:50. doi:10.1186/s40635-019-0263-0
44. Matkovich SJ, Al Khiami B, Efimov IR, et al. Widespread Down-Regulation of Cardiac Mitochondrial and Sarcomeric Genes in Patients With Sepsis. Crit Care Med. 2017;45:407–414. doi:10.1097/CCM.0000000000002207
45. Brusletto BS, Loberg EM, Hellerud BC, et al. Extensive Changes in Transcriptomic “Fingerprints” and Immunological Cells in the Large Organs of Patients Dying of Acute Septic Shock and Multiple Organ Failure Caused by Neisseria meningitidis. Front Cell Infect Microbiol. 2020;10:42. doi:10.3389/fcimb.2020.00042
46. Liu W, Li Y, Zhang Y, et al. Identification of biomarkers and immune infiltration in acute myocardial infarction and heart failure by integrated analysis. Biosci Rep. 2023:43. doi:10.1042/BSR20222552
47. Muller I, Vogl T, Kuhl U, et al. Serum alarmin S100A8/S100A9 levels and its potential role as biomarker in myocarditis. ESC Heart Fail. 2020;7:1442–1451. doi:10.1002/ehf2.12760
48. Wang S, Song R, Wang Z, et al. S100A8/A9 in Inflammation. Front Immunol. 2018;9:1298. doi:10.3389/fimmu.2018.01298
49. Fan J, Shi S, Qiu Y, et al. Analysis of signature genes and association with immune cells infiltration in pediatric septic shock. Front Immunol. 2022;13:1056750. doi:10.3389/fimmu.2022.1056750
50. Mahler M, Meroni PL, Infantino M, et al. Circulating Calprotectin as a Biomarker of COVID-19 Severity. Expert Rev Clin Immunol. 2021;17:431–443. doi:10.1080/1744666X.2021.1905526
51. Schiopu A, Cotoi OS. S100A8 and S100A9: dAMPs at the crossroads between innate immunity, traditional risk factors, and cardiovascular disease. Mediators Inflamm. 2013;2013:828354. doi:10.1155/2013/828354
52. Ehrchen JM, Sunderkotter C, Foell D, et al. The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J Leukoc Biol. 2009;86:557–566. doi:10.1189/jlb.1008647
53. Rochette L, Dogon G, Rigal E, et al. Involvement of Oxidative Stress in Protective Cardiac Functions of Calprotectin. Cells. 2022:11. doi:10.3390/cells11071226
54. Vogl T, Stratis A, Wixler V, et al. Autoinhibitory regulation of S100A8/S100A9 alarmin activity locally restricts sterile inflammation. J Clin Invest. 2018;128:1852–1866. doi:10.1172/JCI89867
55. Gong R, Luo H, Long G, et al. Integrative proteomic profiling of lung tissues and blood in acute respiratory distress syndrome. Front Immunol. 2023;14:1158951. doi:10.3389/fimmu.2023.1158951
56. Della Corte V, Tuttolomondo A, Pecoraro R, et al. Inflammation, Endothelial Dysfunction and Arterial Stiffness as Therapeutic Targets in Cardiovascular Medicine. Curr Pharm Des. 2016;22:4658–4668. doi:10.2174/1381612822666160510124801
57. Liberale L, Ministrini S, Carbone F, et al. Cytokines as therapeutic targets for cardio- and cerebrovascular diseases. Basic Res Cardiol. 2021;116:23. doi:10.1007/s00395-021-00863-x
58. Boyd JH, Kan B, Roberts H, et al. S100A8 and S100A9 mediate endotoxin-induced cardiomyocyte dysfunction via the receptor for advanced glycation end products. Circ Res. 2008;102:1239–1246. doi:10.1161/CIRCRESAHA.107.167544
59. Tousif S, Singh AP, Umbarkar P, et al. Ponatinib Drives Cardiotoxicity by S100A8/A9-NLRP3-IL-1beta Mediated Inflammation. Circ Res. 2023;132:267–289. doi:10.1161/CIRCRESAHA.122.321504
60. Marin-Garcia J, Goldenthal MJ, Moe GW. Mitochondrial pathology in cardiac failure. Cardiovasc Res. 2001;49:17–26. doi:10.1016/s0008-6363(00)00241-8
61. Zhou H, Dai Z, Li J, et al. TMBIM6 prevents VDAC1 multimerization and improves mitochondrial quality control to reduce sepsis-related myocardial injury. Metabolism. 2023;140:155383. doi:10.1016/j.metabol.2022.155383
62. Wang Y, Jasper H, Toan S, et al. Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox Biol. 2021;45:102049. doi:10.1016/j.redox.2021.102049
63. Yang H, Zhang Z. Sepsis-induced myocardial dysfunction: the role of mitochondrial dysfunction. Inflamm Res. 2021;70:379–387. doi:10.1007/s00011-021-01447-0
64. Liu S, Chong W. Roles of LncRNAs in Regulating Mitochondrial Dysfunction in Septic Cardiomyopathy. Front Immunol. 2021;12:802085. doi:10.3389/fimmu.2021.802085
65. Jin Z, Ji Y, Su W, et al. The role of circadian clock-controlled mitochondrial dynamics in diabetic cardiomyopathy. Front Immunol. 2023;14:1142512. doi:10.3389/fimmu.2023.1142512
66. Wang Q, Sun Z, Cao S, et al. Reduced Immunity Regulator MAVS Contributes to Non-Hypertrophic Cardiac Dysfunction by Disturbing Energy Metabolism and Mitochondrial Homeostasis. Front Immunol. 2022;13:919038. doi:10.3389/fimmu.2022.919038
67. Tian J, Zheng Y, Mou T, et al. Metformin confers longitudinal cardiac protection by preserving mitochondrial homeostasis following myocardial ischemia/reperfusion injury. Eur J Nucl Med Mol Imaging. 2023;50:825–838. doi:10.1007/s00259-022-06008-z
68. Vasileiou PVS, Evangelou K, Vlasis K, et al. Mitochondrial Homeostasis and Cellular Senescence. Cells. 2019;8. doi:10.3390/cells8070686
69. Palikaras K, Daskalaki I, Markaki M, et al. Mitophagy and age-related pathologies: development of new therapeutics by targeting mitochondrial turnover. Pharmacol Ther. 2017;178:157–174. doi:10.1016/j.pharmthera.2017.04.005
70. Ambekar T, Pawar J, Rathod R, et al. Mitochondrial quality control: epigenetic signatures and therapeutic strategies. Neurochem Int. 2021;148:105095. doi:10.1016/j.neuint.2021.105095
71. Ashar FN, Zhang Y, Longchamps RJ, et al. Association of Mitochondrial DNA Copy Number With Cardiovascular Disease. JAMA Cardiol. 2017;2:1247–1255. doi:10.1001/jamacardio.2017.3683
72. Tang YS, Zhao YH, Zhong Y, et al. Neferine inhibits LPS-ATP-induced endothelial cell pyroptosis via regulation of ROS/NLRP3/Caspase-1 signaling pathway. Inflamm Res. 2019;68:727–738. doi:10.1007/s00011-019-01256-6
73. Tian C, Liu Y, Li Z, et al. Mitochondria Related Cell Death Modalities and Disease. Front Cell Dev Biol. 2022;10:832356. doi:10.3389/fcell.2022.832356
74. Angajala A, Lim S, Phillips JB, et al. Diverse Roles of Mitochondria in Immune Responses: novel Insights Into Immuno-Metabolism. Front Immunol. 2018;9:1605. doi:10.3389/fimmu.2018.01605
75. Zhao W, Feng H, Sun W, et al. Tert-butyl hydroperoxide (t-BHP) induced apoptosis and necroptosis in endothelial cells: roles of NOX4 and mitochondrion. Redox Biol. 2017;11:524–534. doi:10.1016/j.redox.2016.12.036
76. Cadenas S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic Biol Med. 2018;117:76–89. doi:10.1016/j.freeradbiomed.2018.01.024
77. Santos-Miranda A, Joviano-Santos JV, Ribeiro GA, et al. Reactive oxygen species and nitric oxide imbalances lead to in vivo and in vitro arrhythmogenic phenotype in acute phase of experimental Chagas disease. PLoS Pathog. 2020;16:e1008379. doi:10.1371/journal.ppat.1008379
78. Faria A, Persaud SJ. Cardiac oxidative stress in diabetes: mechanisms and therapeutic potential. Pharmacol Ther. 2017;172:50–62. doi:10.1016/j.pharmthera.2016.11.013
79. Lee SR, An EJ, Kim J, et al. Function of NADPH Oxidases in Diabetic Nephropathy and Development of Nox Inhibitors. Biomol Ther (Seoul). 2020;28:25–33. doi:10.4062/biomolther.2019.188
80. Joseph LC, Kokkinaki D, Valenti MC, et al. Inhibition of NADPH oxidase 2 (NOX2) prevents sepsis-induced cardiomyopathy by improving calcium handling and mitochondrial function. JCI Insight. 2017:2. doi:10.1172/jci.insight.94248
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.