")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 10
Transforming growth factor-β1/Smad3-independent epithelial–mesenchymal transition in type I collagen glomerulopathy
Authors Brodeur AC, Roberts-Pilgrim AM, Thompson KL, Franklin CL, Phillips CL
Received 10 May 2017
Accepted for publication 14 July 2017
Published 31 August 2017 Volume 2017:10 Pages 251—259
DOI https://doi.org/10.2147/IJNRD.S141393
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pravin Singhal
Amanda C Brodeur,1–3 Anna M Roberts-Pilgrim,3 Kimberlee L Thompson,1 Craig L Franklin,4 Charlotte L Phillips2,3
1Department of Biomedical Sciences, Missouri State University, Springfield, MO, USA; 2Department of Child Health, University of Missouri, Columbia, MO, USA; 3Department of Biochemistry, University of Missouri, Columbia, MO, USA; 4Department of Veterinary Pathobiology, University of Missouri, Columbia, MO, USA
Abstract: The glomerulofibrotic Col1a2-deficient mouse model demonstrates glomerular homotrimeric type I collagen deposition in mesangial and subendothelial spaces. In this report, we investigate the role of transforming growth factor β1 (TGF-β1) in myofibroblast activation and epithelial–mesenchymal transition (EMT) in this glomerulopathy. Immunohistochemical analyses of glomerular α-sma, desmin, vimentin, and proliferating cell nuclear antigen demonstrated parietal epithelial cell proliferation and EMT in late stages of the glomerulopathy in the Col1a2-deficient mice. Glomerular TGF-β1 RNA and protein were not elevated in 1- and 3-month-old mice as determined by quantitative reverse transcriptase-polymerase chain reaction and protein immunoassay analyses. To investigate further whether TGF-β1 plays a role in the glomerulopathy outside of the 1- and 3-month time periods, the Col1a2-deficient mice were bred with Smad3 knockout mice. If the glomerular fibrosis in the Col1a2-deficient mice is mediated by the TGF-β1/Smad3 transcription pathway, it was hypothesized that the resultant Col1a2-deficient/Smad3-deficient mice would exhibit attenuated glomerular homotrimer deposition. However, the Col1a2-deficient/Smad3-deficient kidneys were similarly affected as compared to age-matched Col1a2-deficient kidneys, suggesting that homotrimeric type I collagen deposition in the Col1a2-deficient mouse is independent of TGF-β1/Smad3 signaling. Deposition of homotrimeric type I collagen appears to be the initiating event in this glomerulopathy, providing evidence that EMT and myofibroblast activation occur following initiation, consistent with a secondary wound-healing response independent of TGF-β1.
Keywords: renal pathology, glomerulofibrosis, myofibroblast, homotrimeric type I collagen
Introduction
Epithelial–mesenchymal transition (EMT) is a change from an epithelial phenotype to a mesenchymal cell phenotype that occurs during both development and disease. This change is characterized by changes in epithelial protein expression, reorganization of actin protein, basement membrane disruption, and increased cell mobility because of altered cell adhesion.1 EMT is a well-documented phenomenon demonstrated in normal renal embryology and in the renal tubular epithelium of disease models involving renal fibrosis.2–7 EMT has been shown to occur in glomerular epithelial cells in human pauci-immune crescentic glomerulonephritis (PICGN)3,4 as well as in rat models with 5/6 nephrectomy and antiglomerular basement membrane disease.5 Evidence of EMT is seen in renal glomeruli by examining localization and differential expression of mesenchymal proteins such as desmin, vimentin, proliferating cell nuclear antigen (PCNA), and α-smooth muscle actin (α-SMA).6
At the molecular level, there is growing evidence that transforming growth factor β1 (TGF-β1) activation of Smad3-dependent signaling mediates a cascade of fibrotic and EMT responses resulting in renal fibrosis.8–10 TGF-β1 protein levels have been shown to be increased and linked to EMT in mesangial cells prior to the onset of kidney fibrosis.11 TGF-β1 has been found to be upregulated in numerous mouse and human models of glomerular fibrosis and chronic renal disease.12–15 It is also thought to induce podocytopenia, mesangial cell hypertrophy, and extracellular matrix (ECM) production.16 Mechanically, TGF-β1 can be activated by cytoskeleton tension, which occurs as cells progress through EMT.17 Further, TGF-β1 has been shown to be one of the pathways to mesangial cell activation where inflammatory cells are signaled and activated, and is involved in the activation and differentiation of epithelial cells and fibroblasts into myofibroblasts. Because of its involvement with these inflammatory processes, TGF-β is thought to signal other inflammatory mediators such as tumor necrosis factor alpha (TNF-α) and interleukins.16 After phosphorylation by the TGF-β1/TGFβ-type II transmembrane receptor, Smad3 has been shown to mediate the profibrotic activities of TGF-β1.6 This leads to a cellular transition into a myofibroblast-like state, as well as stimulating EMT through a wide variety of mediators,18 all of which result in ECM production by activated cells in vitro and increased type I collagen expression.19–21 Additionally, Smad3 null mice have been shown to be resistant to streptozotocin-induced glomerular fibrosis, bleomycin-induced pulmonary fibrosis, and carbon tetrachloride-induced hepatic fibrosis.22
Type I collagen production and deposition had been demonstrated in many models and types of renal disease.23–25 We have previously shown progressive postnatal deposition of homotrimeric type I collagen, α1(I)3, in the Col1a2-deficient mouse model, initially identified in the glomerular tuft and renal mesangium.26–28 Electron microscopy revealed localization of fibrillar homotrimer between the mesangial matrix and within the subendothelial space, along with podocyte effacement, ultimately resulting in proteinuria.27 Other pathologic changes found in the homotrimeric type I collagen glomerulopathy include parietal epithelial cell hyperplasia, thickening of the capsule basement membrane, and periglomerular fibrosis. The cell type responsible for the collagen production and the mechanism of initiation remain unclear. To further define the mechanism of type I collagen deposition in this mouse model, we have demonstrated that homotrimeric type I collagen synthesis is increased via increases in COL1A1 mRNA, and that its degradation by local matrix metalloproteinases is impaired.27–30 In other mouse models, initiation of glomerular fibrosis and interstitial collagen production has been demonstrated to occur through mesangial cell activation, EMT, and/or migration of interstitial fibroblasts into glomeruli.5,12,13 However, type I collagen production is more commonly seen in secondary stages of wound healing, rather than the initiation of renal disease.
In this study, we sought to further characterize the cellular and molecular mechanisms responsible for deposition of homotrimeric type I collagen in the glomerulus. We demonstrate by immunohistochemical staining of desmin, vimentin, PCNA, and α-SMA, mesangial cell activation and parietal cell EMT in affected glomeruli following the onset of the glomerulopathy. Further, we sought to determine if the most common inducer of EMT, TGF-β1 signaling via Smad3, was responsible for cellular transdifferentiation and homotrimeric type I collagen deposition. Our studies indicate that the type I collagen glomerulopathy is not mediated by the TGF-β1/Smad3 pathway. We demonstrate TGF-β1/Smad3-independent initiation of the homotrimeric type I collagen glomerulopathy in the Col1a2-deficient mice through evaluation of steady-state TGF-β1 mRNA by quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR), TGF-β1 protein localization by immunohistochemistry (IHC), TGF-β1 protein quantification using array technology, and abrogation of Smad3 signaling using Smad3 knockout mice.
Materials and methods
Ethics statement
All animal care conformed to the National Institutes of Health Guide for Care and Use of Laboratory Animals and was approved by the University of Missouri Animal Care and Use Committee (an AAALAC accredited animal facility), Columbia, MO, USA (Protocol Registry Number 3579; Animal Welfare Assurance Number A3394–01).
Animals – Col1a2-deficient
Heterozygous B6C3Fe a/a-Col1a2oim/J (Col1a2-deficient +/−) mice were purchased from the the Jackson Laboratory (Bar Harbor, ME, USA), and bred and genotyped as previously published to generate wildtype (+/+), heterozygous (+/−), and Col1a2-deficient (−/−) animals.31 Animals were fed (Purina 5008 Formulab Diet; Purina Mills Inc., Richmond, IN, USA) ad libitum. Col1a2-deficient mice are also known as the osteogenesis imperfecta murine model, or oim.
Animals – Col1a2-deficient/Smad3-deficient
Homozygous male B6C3Fe a/a-Col1a2oim/J (−/−) mice, purchased from Jackson Laboratories, were mated with a heterozygous 129-Smad3tm1Par/J female mouse (Smad3+/−) generously provided by Dr. Lillian Maggio-Price. Breeding pairs were generated from F1 offspring. Smad3 genotypes were determined by PCR as previously described.32 Genotypes were denoted Col1a2-deficient genotype/Smad3-deficient genotype. Kidneys were harvested at 1 month of age, formalin-fixed, embedded in paraffin, longitudinally sectioned (5 µm), and analyzed by light microscopy (n=6 [+/+, +/+], n=6 [+/+, Smad3+/−], n=2 [+/+, Smad3−/−]; n=7 [+/−, +/+], n=14 [+/−, Smad3+/−], n=10 [+/−, Smad3−/−]; n=6 [−/−,+/+], n=18 [−/−, Smad3+/−], n=9 [−/−, Smad3−/−]).
Histomorphometry
Kidneys were harvested at 1 week and 1 month of age, formalin-fixed, embedded in paraffin, and longitudinally sectioned (5 µm). Sections were stained with picrosirius red or used for IHC. Kidneys were blindly assigned lesion scores as previously reported.27 Briefly, G1 – mild lesions, <50% of glomeruli affected; G2 – moderate lesions, <50% of glomeruli affected; G3 – moderate lesions, >50% glomeruli affected; G4 – severe lesions, >50% of glomeruli affected.27
Glomerular isolation
Wildtype (+/+) and (−/−) mice were aged 1 and 3 months and anesthetized prior to kidney perfusion.33 Perfusion of 1× PBS and 8×107 tosylactivated Dynabeads® magnetic beads, deactivated prior to use according to the manufacturer’s instructions, were perfused through the body via the right ventricle of the heart. Following perfusion, kidneys were removed, weighed, and minced followed by thorough mixing with digestion solution (1 mg/mL collagenase A [Invitrogen Corporation, Carlsbad, CA, USA], 100 units/mL DNase [Invitrogen Corporation], and Hanks Balanced Salt Solution [HBSS] [Invitrogen Corporation]) and incubation at 37°C for 30 minutes. The digested slurry was sieved twice with 100 micron cell strainers (BD Biosciences, San Jose, CA, USA) and addition of HBSS, followed by centrifugation at 1500 rpm for 15 minutes. Pellet was resuspended in HBSS and placed onto a magnetic particle concentrator for 1 minute to separate glomeruli from extraneous tissue for a total of 5 washes. Remaining glomeruli were resuspended in HBSS and assessed for purity and yield with a hemocytometer, followed by snap-freezing and storage at −80°C.
Quantitative reverse transcriptase-polymerase chain reaction
Snap-frozen glomeruli from 1-month-old (n=10 [−/−] [lesion score G3–4], n=6 [+/+] [lesion score G0]) and 3-month-old (n=8[−/−] [lesion score G1–4], n=10[+/+] [lesion score G0]) mice were homogenized in TRIzol Reagent (Invitrogen Corporation) using a TissueLyser homogenizer (QIAGEN, Valencia, CA, USA), or RNeasy Kit (QIAGEN) and total RNA isolated according to the manufacturer’s protocol. Total glomerular RNA was transcribed following the manufacturer’s protocol (SuperScript First-Strand Synthesis or VILO; Invitrogen Corporation), and qRT-PCR amplification and standard curve generation were performed as previously described. Primer sequences for HPRT34 and TGF-β135 have been previously reported. TGF-β1 transcripts were normalized to HPRT copy number.
Searchlight protein assay
Snap-frozen glomeruli from 1-month-old (n=11 [−/−], and n=9 [+/+]) and 3-month-old (n=5 [−/−], n=5 [+/+]) wildtype and Col1a2-deficient mice were thawed on ice, and protease activity was inhibited by the addition of phenylmethyl sulfonyl fluoride to a final concentration of 2 mM. Glomeruli were sonicated in an ice waterbath for 10 minutes to disrupt the glomerular unit, then incubated for 1 hour at room temperature with agitation (200 rpm) to allow antigen–protein binding to piezoelectrically adhered TGF-β1 antigens in a 96-well plate format followed by washing for removal of unbound proteins. Biotinylated detection antibodies also specific for TGF-β1 detection were added to the 96-well plate and incubated for 30 minutes, then a 30-minute incubation with streptavidin horseradish peroxidase (HRP) conjugate and imaged by SuperSignal ELISA Femto Chemiluminescence substrate. Protein quantitation was determined by Thermo Scientific SearchLight multiplex assay (Pierce Scientific, Rockford, IL, USA) according to the manufacturer’s procedures.
Immunohistochemistry
Formalin-fixed kidneys harvested from 1-week- and 1-month-old mice were embedded in paraffin and sectioned at 5 µm. α-sma IHC primary antibody at 1:200 (mouse monoclonal anti-sma [2547]; Sigma, St. Louis, MO, USA) was used with the Mouse on Mouse kit (M.O.M., Vector Laboratories, Burlingame, CA, USA). TGFβ-R1 IHC using 1) rabbit polyclonal anti-TGFβ-RI primary antibody at 1:50 (sc-398; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and 2) chicken polyclonal anti-TGF-β1 primary antibody at 1:100 (AB-101-NA; R & D Systems, Minneapolis, MN, USA). Antibody detection was done using an avidin–biotin–peroxidase complex method (kit PK-4002; Vector Laboratories) with diaminobenzidine-H2O2 (Sigma) and counterstained with hematoxylin. Desmin IHC used heat-induced epitope retrieval in 1× target retrieval solution (TRS) (Dako, Carpinteria, CA, USA) followed by incubation with rabbit polyclonal anti-desmin (ab15200) primary antibody at 1:200 (Abcam, Cambridge, MA, USA) and EnVision + System-HRP-labeled polymer anti-rabbit (K4003) detection of HRP-labeled polymers conjugated with anti-rabbit antibodies (Dako). Vimentin IHC used heat-induced epitope retrieval in 1× TRS followed by incubation with goat polyclonal anti-Vimentin (c-20, sc7557) primary antibody at 1:50 (Santa Cruz Biotechnology) and biotinylated rabbit anti-goat secondary antibody at 1:400. PCNA IHC used heat-induced epitope retrieval in 1× TRS followed by incubation with mouse anti-PCNA (M0879) primary antibody at 1:400 (Dako) and biotinylated rabbit anti-mouse secondary antibody at 1:200. TGF-β1 IHC (Promega) used heat-induced epitope retrieval in 1× TRS followed by incubation with rabbit polyclonal anti-TGF-β1 (G1221) primary antibody at 1:50 (Promega) and EnVision + System HRP-labeled polymer anti-rabbit (K4003) detection of HRP-labeled polymers conjugated with anti-rabbit antibodies (Dako). Slides were then incubated in a streptavidin HRP conjugate, and 3,3′ diaminobenzidine tetrahydrochloride and counterstained with hematoxylin. Desmin, vimentin, TGF-β1 (Promega), and PCNA staining was done on a Dako Autostainer Universal Staining System.
Statistics
Statistical analyses were completed using SAS (SAS Institute Inc., Cary, NC, USA). The TGF-β1 RT-PCR and protein data were analyzed by analysis of variance as a completely randomized design in which genotype and age were arranged as a 3×2 factorial. Data are presented as mean and standard error. Differences were considered to be significant at p<0.05.
Results
α-sma, desmin, vimentin, and PCNA
Renal localization of α-sma, desmin, vimentin, and PCNA was determined by immunohistochemical evaluation of kidneys from wildtype (+/+) and Col1a2-deficient (−/−) mice to determine if mesangial cells were proliferating and becoming activated (Figure 1). At 1 week of age +/+ and −/− kidneys demonstrated similar staining patterns for α-sma, desmin, vimentin, and PCNA across genotypes. By 1 month of age, α-sma-, desmin-, and vimentin-positive staining was present in the parietal epithelial cells of Col1a2-deficient kidneys in contrast to age-matched wildtype kidneys. Further, severely affected mice demonstrated increased α-sma, desmin, vimentin, and PCNA antibody binding to mesangial cells.
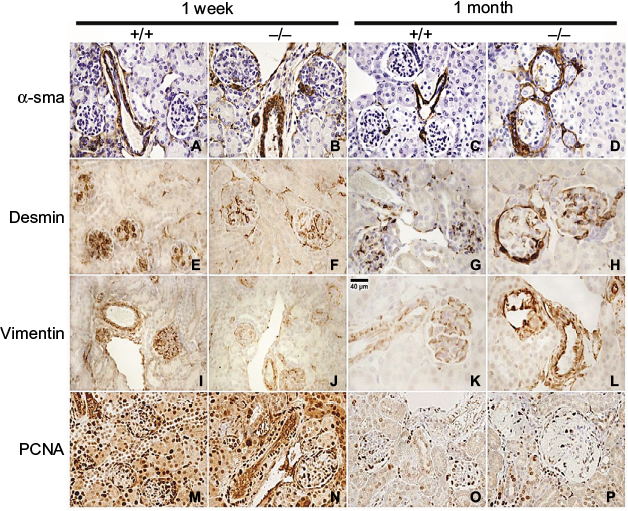 | Figure 1 Col1a2-deficient (−/−) glomeruli demonstrate PEC EMT at 1 month of age. Notes: Immunostaining of 1-week and 1-month +/+ and −/− kidneys with α-sma, desmin, vimentin, and PCNA. Immunostaining at 1 week of age demonstrates glomerular α-sma (A, B), desmin (E, F), vimentin (I, J), and PCNA (M, N) in both +/+ and −/− within the same regions, consistent with developmental expression. α-sma is seen in the vascular pole of +/+ glomeruli (C), in contrast to −/− glomeruli (D), which show α-sma staining in PECs and in the mesangium. At 1 month of age,+/+ kidneys demonstrate desmin (G) and vimentin (K) staining in podocytes, while −/− glomeruli demonstrate intense desmin (H) and vimentin (L) staining in PECs and a redistribution of positive staining in podocytes to the periphery. PCNA-positive staining was seen in +/+(O) and −/− (P) glomeruli at 1 month of age; however, there was a decrease in intraglomerular staining and an increase in PEC positivity as compared to +/+ glomeruli. Abbreviations: EMT, epithelial–mesenchymal transition; PCNA, proliferating cell nuclear antigen; PEC, parietal epithelial cell. |
α-sma and vimentin,36 markers of a mesenchymal phenotype, were found to be differentially expressed in the Col1a2-deficient kidneys relative to wildtype kidneys (Figure 1). In wildtype mice, α-sma was expressed in the vascular pole of the glomeruli, whereas in Col1a2-deficient mice, α-sma staining was seen in the parietal epithelial cells and less so in the mesangium of severely affected mice. Vimentin demonstrated a characteristic staining profile in wildtype kidneys, staining positive in glomerular epithelial cells, or podocytes, and demonstrating an absence of staining in parietal epithelial cells. In Col1a2-deficient kidneys, the opposite was observed, and vimentin stained the parietal epithelial cells intensely positive and was absent or masked in the glomerular epithelial cells.
Desmin, a marker of myogenic properties in glomerular epithelial cells,36 was found to be differentially localized in the Col1a2-deficient mouse kidney at 1 month of age (Figure 1). Desmin-positive staining exhibited a homogeneous pattern in wildtype glomerular epithelial cells at 1 month of age. However, at 1 month of age, Col1a2-deficient kidneys demonstrated more intense staining of the parietal epithelial cells, in addition to the presence of desmin-positive staining in glomerular epithelial cells in younger mice.
PCNA-positive staining was homogeneous throughout the glomeruli of wildtype kidneys (Figure 1). There was decreased PCNA-positive staining within the glomerulus of Col1a2-deficient kidneys, whereas the parietal epithelial cells were found to be strongly positive.
TGF-β1: molecular approach
qRT-PCR was performed on total RNA isolated from glomeruli of 1- and 3-month-old wildtype (+/+) and Col1a2-deficient (−/−) mice and confirmed that TGF-β1 steady-state mRNA is not elevated in Col1a2-deficient glomeruli relative to wildtype glomeruli (Figure 2A). On the contrary, TGF-β1 mRNA copy number was found to be increased in wildtype (+/+) glomeruli relative to Col1a2-deficient glomeruli.
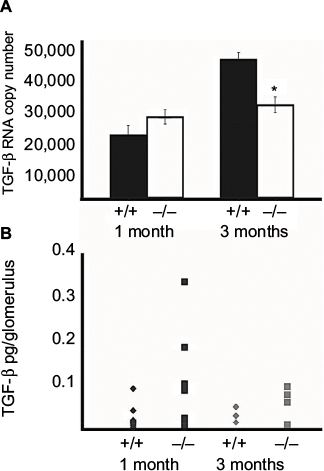 | Figure 2 TGF-β1 steady-state mRNA and protein are not upregulated in homotrimeric type I collagen glomerulopathy. Notes: (A) Quantitative RT-PCR demonstrates that TGF-β1 copy number is not significantly different in glomerular isolates of 1-month-old wildtype (+/+) and Col1a2-deficient (−/−) mice. However, by 3 months of age the TGF-β1 copy number is reduced in Col1a2-deficient glomerular isolates as compared to wildtype glomerular isolates (*p≤ 0.05). (B) Although protein immunoassay demonstrated no significant difference in picograms of TGF-β1 protein per glomerulus in wildtype (+/+) and Col1a2-deficient (−/−) glomeruli at both 1 month and 3 months of age, Col1a2-deficient (−/−) glomeruli at 1 month of age exhibited a greater variability in TGF-β1 protein per glomerulus. Abbreviations: TGF-β1, tumor growth factor β1; RT-PCR, reverse transcriptase-polymerase chain reaction. |
TGF-β1: biochemical approach
To determine, by biochemical approaches, if TGF-β1 is involved in mediation of the type I collagen glomerulopathy as it is in other mouse models of glomerulosclerosis, localization by IHC and quantitation by immunoassay analysis were performed (Figure 2B). IHC on frozen and paraffin-embedded kidneys from 1-month-old mice did not localize TGF-β1 to the glomeruli in wildtype, heterozygous, or Col1a2-deficient kidneys (data not shown). Examination of TGF-β1 protein levels in wildtype and Col1a2-deficient glomeruli by array analysis confirmed that there were no significant differences in TGF-β1 protein at 1 and 3 months of age.
TGF-β1: genetic approach
By biochemical and molecular approaches, differences in TGF-β1 were not found between genotypes at 1 and 3 months of age. However, the glomerulopathy initiates prior to 1 week of age. Therefore, we sought to determine if TGF-β1/Smad3 signaling was involved in earlier, prior to 1 week, disease processes. Histologic examination of kidneys from Col1a2-deficient/Smad3-deficient mice demonstrated that the loss of Smad3 signaling does not alter the development of the type I collagen glomerulopathy (Figure 3). Mice that are deficient in the a2 chain of type I collagen, making only homotrimer, demonstrate lesion scores and severity with no significant difference when compared to the lesion scores of those that are deficient in both the a2 chain and Smad3 signaling. Col1a2-deficient/Smad3-deficient animals develop the type I collagen glomerulopathy and demonstrate severe deposition as early as 1 month of age, as signified by lesion scores of 4 at 1 month of age. The average lesion score for 1-month Col1a2-deficient/wildtype (−/−, +/+) animals is 3.5±0.33, whereas the average lesion score for 1-month Col1a2-deficient/Smad3-deficient (−/−, Smad3−/−) animals is 3.22±0.27, showing no statistical difference.
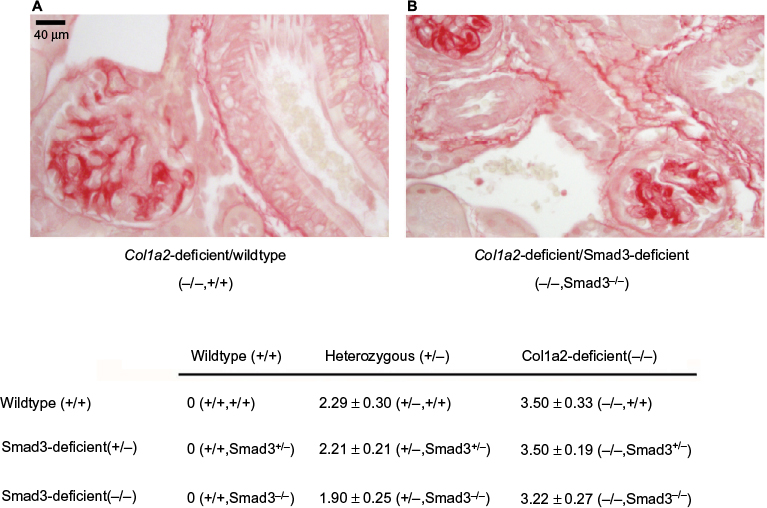 | Figure 3 Col1a2-deficient/Smad3-deficient glomeruli demonstrate homotrimeric type I collagen deposition. Notes: (A) 1-month Col1a2-deficient glomerulus demonstrating fibrillar collagen deposition upon staining with picrosirius red. (B) 1-month Smad3-deficient glomeruli demonstrating similar intensity of picrosirius red staining. Mean lesion scores for 1-month Col1a2-deficient/Smad3-deficient animals are provided in the table and demonstrate no significant differences. |
Discussion
Our work demonstrates similar findings as those seen in PICGN human and rat models, confirming that EMT is occurring.4,37,38 At 1 week of age, we saw similar immunostaining patterns for α-sma, desmin, and vimentin in +/+ and −/− kidneys, which was likely due to the role of these proteins in renal development, as glomerular maturation continues postnatally and nears completion at 21 days of postnatal life.39,40 Parietal epithelial cells in Col1a2-deficient kidneys demonstrate de novo expression of α-sma and acquisition of vimentin expression, suggesting that EMT is participating in the type I collagen glomerulopathy. Our results also suggest that parietal epithelial cells are proliferating because of increased PCNA localization, similar to that characteristic of crescentic glomerulosclerosis. It has also been shown that glomerular epithelial cells demonstrate increased and more intense desmin immunostaining in animals that demonstrate proteinuria and foot process effacement,36 as is seen in Col1a2-deficient animals. However, our work indicates that the EMT and type I collagen deposition in the renal glomeruli of Col1a2-deficient animals are independent of TGF-β1/Smad3 signaling seen in other models.
Glomerular endothelial cell damage is present in various disease states such as preeclampsia,41 diabetic nephropathy,42,43 and hypertension.44,45 Increased glomerular capillary pressure has been shown to result in ECM and type I collagen expression mediated by TGF-β1.46,47 It can be inferred from previous studies demonstrating altered aortic vascular biomechanics in the Col1a2-deficient mice that the glomerular vasculature may exhibit altered biomechanical properties.48,49 One can then postulate that these biomechanical differences may allow for differences in response to mechanostress in the glomerulus and thus may result in homotrimeric type I collagen deposition leading to podocyte effacement, proteinuria, and a secondary wound-healing response including myofibroblast activation and EMT. However, our findings also suggest that such stresses if present are not mediated by TGF-β1 through Smad3 in this model, as demonstrated by molecular, biochemical, and genetic approaches. This may be, in part, due to TGF-β1/Smad3-induced ECM production occurring by way of Smad3 binding to the COL1A2 promoter, which, while functional in Col1a2-deficient mice, may not increase homotrimeric type I collagen production.50
In light of the absence of TGF-β1/Smad3 regulation of glomerular homotrimeric type I collagen deposition, other mediators of fibrosis (such as fibroblast growth factor, connective tissue growth factor [CTGF], platelet-derived growth factor [PDGF], and bone morphogenetic protein 73,46,51,52), inflammation (TNF-α),16,53 and Smad-3-independent TGF-β1 signaling (such as Ras/Raf, extracellular signal-regulated kinase-mitogen-activated protein kinase, and nuclear factor κB) remain to be investigated.46,52 TNF-α has been shown to be released by mesangial cells upon exposure to certain pathogens.54 Some studies have suggested that mesangial-derived TNF-α is responsible for podocyte effacement leading to proteinuria55 as well as complete podocyte loss.56 Recent literature indicates that CTGF and PDGF are potential candidates for investigation.51,57–59 Incubation of cultured rat mesangial cells with recombinant human CTGF resulted in 64% increase in type I collagen secretion, an increase greater than that seen with incubation in the presence of TGF-β1. Further, CTGF transcripts were increased by twofold in cultured rat mesangial cells that underwent cyclic mechanical strain.60 In vivo transfection of PDGF into the kidney of Sprague–Dawley rats demonstrated increased immunostaining with PCNA, as well as increased cell proliferation, type I collagen mRNA expression, and reducible collagen cross-links in connective tissues (tendon, skin), and was postulated to be involved in late stages of wound healing without induction of inflammation.57 A PDGF-D knockout mouse demonstrated a reduction in the severity of renal fibrosis in the Unilateral Ureteral Obstruction (UUO) mouse model and in a unilateral ischemia–reperfusion injury model.58 An additional study demonstrated that renal fibrosis due to UUO could be prevented in PDGF-C knockout or neutralization.62 Further, inhibition of PDGFR-β by an interferon-γ-peptidomimetic decreased collagen expression and attenuated renal interstitial fibrosis in the mouse UUO fibrotic model.61
In summary, our findings suggest that deposition of homotrimeric type I collagen in the glomeruli results in a secondary wound-healing response through parietal cell hyperplasia, as well as EMT and mesangial cell activation in severely affected animals. We also demonstrate that the TGF-β1/Smad3 signaling pathway, a known mediator of glomerular fibrosis, is not likely a primary mediator of the type I collagen glomerulopathy seen in Col1a2-deficient mice. Further investigation into additional initiators of type I collagen deposition and glomerular fibrosis, including TNF-α, PDGF, and CTGF, may provide additional insights into the initiation of the type I collagen glomerulopathy.
Acknowledgments
The authors are grateful to Dr. Lillian Maggio-Price for generously providing the 129-Smad3tm1Par/J mouse; to Stephanie Carleton, Giedre Turner, and Beth Livingston for assistance in genotyping and necropsy; to Dr. Matt Myles for his assistance in obtaining the RT-PCR data; to Jill Gruenkemeyer, Jan Adair, and Bo Cowan for their assistance in immunostaining; to Howard Wilson for assistance in generating the digital artwork depicted; to Lauren Hibler for assistance in lesions scoring; and to Dr. Mark Ellersieck for assistance with statistical analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.