")
Back to Journals » International Journal of Nanomedicine » Volume 19
Revolutionizing Antiviral Therapeutics: Unveiling Innovative Approaches for Enhanced Drug Efficacy
Authors Megantara S , Rusdin A, Budiman A, Shamsuddin S, Mohtar N, Muchtaridi M
Received 14 November 2023
Accepted for publication 29 February 2024
Published 20 March 2024 Volume 2024:19 Pages 2889—2915
DOI https://doi.org/10.2147/IJN.S447721
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Eng San Thian
Sandra Megantara,1,2 Agus Rusdin,3 Arif Budiman,3 Shaharum Shamsuddin,4 Noratiqah Mohtar,5 Muchtaridi Muchtaridi1,2,6
1Department of Pharmaceutical Analysis and Medicinal Chemistry, Faculty of Pharmacy, Universitas Padjadjaran, Sumedang, 45363, Indonesia; 2Research Collaboration Centre for Theranostic Radio Pharmaceuticals, National Research and Innovation Agency (BRIN), Sumedang, 45363, Indonesia; 3Department of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, Universitas Padjadjaran, Sumedang, 45363, Indonesia; 4School of Health, Universiti Sains Malaysia, Penang, 11800, Malaysia; 5School of Pharmaceutical Sciences, Universiti Sains Malaysia, Penang, 11800, Malaysia; 6Functional Nano Powder University Center of Excellence (FiNder U CoE), Universitas Padjadjaran, Sumedang, 45363, Indonesia
Correspondence: Sandra Megantara, Tel +62-813-2066-3583, Email [email protected]
Abstract: Since the beginning of the coronavirus pandemic in late 2019, viral infections have become one of the top three causes of mortality worldwide. Immunization and the use of immunomodulatory drugs are effective ways to prevent and treat viral infections. However, the primary therapy for managing viral infections remains antiviral and antiretroviral medication. Unfortunately, these drugs are often limited by physicochemical constraints such as low target selectivity and poor aqueous solubility. Although several modifications have been made to enhance the physicochemical characteristics and efficacy of these drugs, there are few published studies that summarize and compare these modifications. Our review systematically synthesized and discussed antiviral drug modification reports from publications indexed in Scopus, PubMed, and Google Scholar databases. We examined various approaches that were investigated to address physicochemical issues and increase activity, including liposomes, cocrystals, solid dispersions, salt modifications, and nanoparticle drug delivery systems. We were impressed by how well each strategy addressed physicochemical issues and improved antiviral activity. In conclusion, these modifications represent a promising way to improve the physicochemical characteristics, functionality, and effectiveness of antivirals in clinical therapy.
Keywords: antiviral drugs, current modifications, drug delivery system, physicochemical properties
Introduction
Virus infection is one of the diseases that causes most death in the last decade, after cardiovascular and cancer diseases. In 2020, virus infection had become the first cause of death when the coronavirus pandemic started to attack the world at the end of the year 2019. There were about 150 million cases of prevalence with the fatality rate of 7.5% caused by this coronavirus disease. In addition, the Human Immunodeficiency Virus (HIV), Hepatitis A Virus (HAV), Hepatitis B Virus (HBV), Herpes Simplex Virus (HSV), and Influenza Virus (IV) are still having a high prevalence of cases.1
Vaccination and the use of immunomodulatory agents are valuable options for preventing and managing coronavirus infections. However, they do not guarantee immunity against the virus. Antiviral and antiretroviral drugs are currently the main therapies used for the treatment of many viral diseases, and in some cases, adjuvant therapy is also added to enhance their effectiveness.2
There are several factors that can render drug therapy ineffective, including Multiple Drug Resistant (MDR) strains, virus mutations, and the physicochemical characteristics of drugs, such as poor water solubility and non-selective virus targeting. Furthermore, synthetic drugs can have many side effects, which can reduce the patient’s quality of life.
Considering the limitations, scientists have been trying several approaches to improve the physicochemical characteristics of the drugs, including structure modification and utilization of drug delivery systems.3
In order to address the physicochemical aspects of drug therapy, including solubility, selectivity, and activity, scientists have successfully employed physical and chemical modification strategies.4 Among the most explored methodologies are nanoparticle drug delivery systems including nanostructured lipid carriers, solid lipid nanoparticle, solid lipid nanoparticle, and hybrid nanoparticle.5–9 Also, others type of modification like liposomes, cocrystals, solid dispersions, salt formation, and structural modification. Numerous studies pertaining to the development of antiviral drugs have been published. However, existing discussions are limited to specific modifications, such as nanoparticle and metal-based modifications.10–12 Currently, there is an absence of comprehensive literature evaluations that systematically analyze, summarize, and compare various approaches within this research domain.13–18
Based on these considerations, the authors are motivated to write an article review on the current developments of antiviral drugs aimed at resolving physicochemical problems and improving their efficacy. This review will provide information for the basic and primary consideration of future drug design and development to enhance the available treatment of viral infections. We posit that our review will be the inaugural examination to explicitly address and draw comparisons among contemporary successful techniques, encompassing chemical and physical modifications, aimed at enhancing the efficacy of antiviral drugs.
Materials and Methods
This review is based on published journals obtained from Scopus, PubMed, and Google Scholar databases using specific keywords such as “nanoparticle antiviral”, “liposome antiviral”, “cocrystal antiviral”, “solid dispersion antiviral”, “salt modification antiviral”, and “structure modification antiviral”. In the primary discourse, we excluded subjective viewpoints, reviews, and tangential subjects. The database search is limited to recent journal publications from 2019 to 2023. The methodology flowchart is illustrated in Figure 1. All the literature sources are used as references for the introduction chapter, while some articles are used for discussion before the main topic, including the current state of nanoparticles development for treating various diseases and the primary discussion on the development of nanoparticles for antiviral drugs.
 |
Figure 1 Flowchart of the Methodology. |
Viruses Definition
Viruses, as per the International Committee on Taxonomy of Viruses (ICTV), are tangible entities resulting from biological evolution and genetics. ICTV categorizes each virus into a single species.19
Contrary to conventional views, Morgan (2016) proposes “radical pluralism” as an alternative strategy for classifying viruses. This non-hierarchical approach accommodates the impact of horizontal genetic transfer on viral evolution, enabling viruses to belong to multiple categories. This flexibility aids in predictions and directs attention to often overlooked biological entities within a non-hierarchical framework.20
Calisher (2016) posits that viruses are unique entities with distinct traits, including the ability to replicate, infect cells, and possess genetic material. Morgan (2016) emphasizes the adaptability and evolution of viruses, highlighting their communication and co-evolution with biological hosts.21,22
Virus Classification
The International Committee on Taxonomy of Viruses (ICTV), formed by the International Union of Microbiological Societies (IUMS), oversees the taxonomy of viruses. ICTV’s objectives encompass establishing universally recognized information on virus taxonomy, naming taxa, communicating taxonomic decisions to the global virology community, and maintaining a list of viral names. Unlike other biological classification systems, ICTV governs and approves the development of viral taxa.19
The hierarchical structure of virus taxonomy consists of order, family, subfamily, genus, and species, with “species” being the lowest rank and “order” the highest. As of 2018, there are eleven defined categories at the “order” level, each grouping various viral families. Examples include Mononegavirales (9 families), Caudovirales (4 families), Herpesvirales (3 families), Ligamenvirales (2 families), Nidovirales (4 families), Ortervirales (5 families), Picornavirales (7 families), and Tymovirales (5 families). Additionally, 86 families are not specifically assigned to any particular group at the “order” level.23
There are many different families of viruses that can cause tumors in humans, including the RNA (Retroviridae and Flaviviridae) and DNA (Hepadnaviridae, Herpesviridae, and Papillomaviridae) virus families.24 Both types encode oncoproteins that can or cannot focus regulatory systems found in host cells based on their genetic constructions.25 The genome of the host cell can be altered by viruses with DNA origins. RNA viruses, on the other hand, are primarily cytoplasmic and cannot be incorporated into the host cell’s genome.21
Retroviruses like human T-cell leukemia virus-1 (HTLV-1), human immunodeficiency virus-1 (HIV-1), and flaviviruses like hepatitis C virus (HCV) are among the RNA viruses that cause cancers.26 The following DNA viruses are among those that induce malignancies in their natural hosts: Hepatitis B virus (HBV), human papillomavirus (HPV), Epstein-Barr virus (EBV), and human herpesvirus 8 or sometimes referred to as Kaposi sarcoma-associated herpesvirus (KSHV),27 and Merkel cell polyomavirus (MCPV).28 While HBV, HPV, and MCPV integrate into the host genome and participate in various carcinogenesis pathways depending on the viral integration region, EBV and HHV-8 do not.3
The molecular mechanisms behind the cellular changes brought on by viruses have been clarified by certain investigations on DNA viruses, such as adenoviruses, polyomaviruses, and papillomaviruses. Despite their divergent evolutionary paths, these viruses’ transformation activities are strikingly similar, highlighting their shared requirement to utilize the host’s replication apparatus for efficient viral reproduction.27
Both DNA and RNA viruses belong to a large number of families that collectively make up the group of tumoral viruses. They produce several kinds of oncoproteins that can either target or ignore host cell regulatory mechanisms. Understanding how viral oncogenes alter the expression of growth-promoting factors has revealed new information about the fundamental mechanisms underlying the genesis of cancer.29 An agent, such as a virus that causes cancers in human, cannot be established or eliminated based on any apparent molecular rule. Furthermore, virtually all viruses that cause tumors resemble viruses that do not cause cancer in human.30
Virus Life Cycle
The process of adhesion, virus entry into a host cell, virus reproduction inside the cell, and finally the release of progeny particles are typically referred to as the “life cycle” of a virus.31,32 The primary process of any virus’ infectivity is cellular entrance. The key chemicals in the identification of target cells that control their cellular tropism and species-specific characteristics are found on their surface.33
Nowak et al (2001) outline eight crucial events in the viral life cycle: (i) viral adhesion to the host cell; (ii) invasion of the cell by the virus or its genetic material; (iii) uncoating of the viral genetic material; (iv) synthesis of viral proteins manipulating host cells; (v) replication of the viral genome by the host cell machinery; (vi) production of additional viral proteins for the capsid.34,35 The visual depiction of the life cycle of the virus is observable in Figure 2.
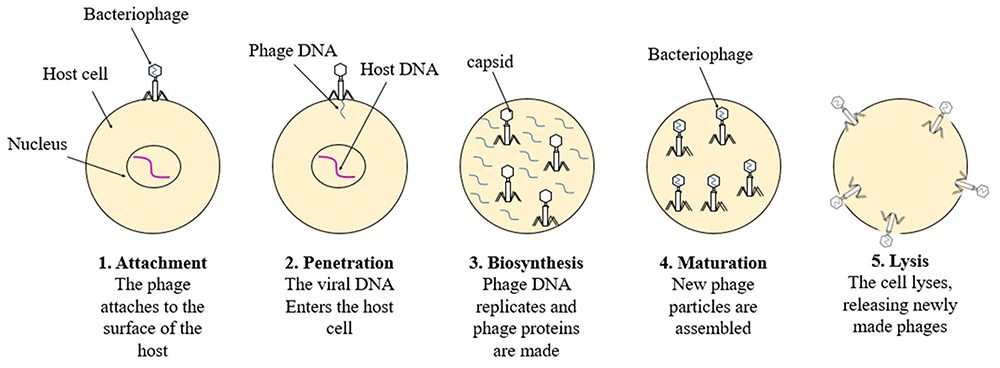 |
Figure 2 Virus Life Cycle. |
The order of phases in a virus’s reproduction cycle might vary greatly between different viruses. These could affect how quickly they kill cells and spread, which would affect their ability to treat cancer. Each virus has a different number of genes and a different level of complexity in how they are regulated. For many viruses, the transport of viral nucleic acids to the nucleus is necessary in order to utilize the host cell’s transcriptional machinery and genome amplification.36
Viral nucleic acids that are deposited in the cytoplasm or nucleus of aberrant cells during infection serve as strong stimulators of immune response pathways. A protein must meet two requirements in order to be designated as a viral DNA detector. The first requirement is for it to physically engage with the viral DNA; the second is for it to activate the innate immune system, which entails inducing the release of interferons, chemokines, and/or cytokines.37 Inflammasomes recognize PAMPs, such as double-stranded DNA, in response to a viral infection.21 The PRRs are responsible for detecting nucleic acids, and these units can also detect PAMPs and DAMPs.38
In the case of viruses that cause disease, the infectious process is metaphorically shown as a conflict between a host and a virus, with the assumption that the virus, as such, is capable of inventing new tactics and methods to get past the host’s defenses. However, in accordance with van Regenmortel and colleagues (2016), the viral enzyme’s reverse transcriptase’s error-prone activity is the only source of stochastic mutations that provide the virus the potential to subvert the immune system.39 According to Crow and colleagues (2015), the dynamic equilibrium between host defense and viruses’ capacity to circumvent these mechanisms affects how effective the immune response is. One method employed by viruses to suppress immune signals is the sequestration of host antiviral components as well as their destruction.25,40,41 Tumor viruses affect practically all important signaling pathways, including MAP kinases, JAK-STAT, TGF-, NF-kB, Notch, TNF, Wnt, and Hedgehog, through their oncoproteins and other regulatory molecules. Tumor viruses may alter these pathways to foster a climate favorable for their replication and to encourage host cells to aggressively divide and proliferate. In most cases, viral oncoproteins rewire host cells by capturing and recycling the host transcriptional networks’ regulatory elements. MAP kinases are active in the presence of cancer and are triggered in response to stress or growth factors (ERK, JNK, and p38).42
Antiviral Drugs
Anti Non-Retroviral Drugs
Antiviral medications treat viral infections specifically. Viruses cause numerous diseases, including cancer and various health issues in humans, plants, and animals. Notable viral illnesses include measles, mumps, smallpox, chickenpox, influenza, poliomyelitis, and yellow fever. Viruses contain either DNA or RNA, but not both. Current antiviral drugs, such as amantadine, vidarabine, trifluridine, idoxuridine, aciclovir, ribavirin, and zidovudine, act by inhibiting viral DNA synthesis. These drugs enhance cell resistance to viruses (interferons), block virus entry into cells (amantadine), and hinder nucleic acid synthesis. Table 1 outlines various non-retroviral antiviral medicines.31
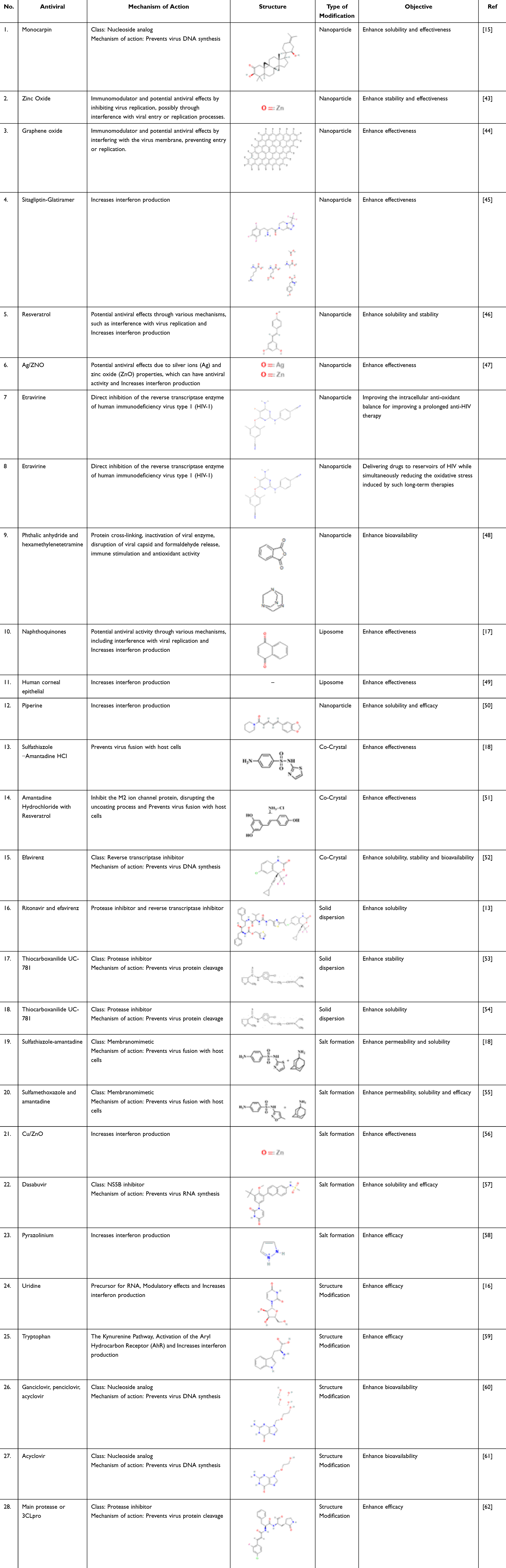 |
Table 1 Current Development of Antiviral Drugs |
Antiretroviral Drugs
As of early 2014, the US Food and Drug Administration (FDA) had approved 28 antiretroviral medications across six mechanistic classes. Newer drugs, with increased potency, reduced toxicity, fewer pills, and less frequent administration, have largely replaced older ones. This multifaceted pharmacotherapeutic array enables the tailoring of therapeutic interventions. Despite the efficacy of recent antiretrovirals, HIV persists in sanctuary and reservoirs, leading to viral rebound even after brief treatment interruptions. Continuous medication is necessary for viral suppression, posing challenges for asymptomatic HIV patients due to potential drug resistance with intermittent compliance. An article discusses antiretroviral therapy objectives, principles, drug regimen selection, and the pharmacology of widely prescribed antiretroviral medications.31
Antiviral medications are chemical substances that, through a variety of mechanisms of action, have the ability to prevent virus infection from growing.
Anti-Viral Drug Classifications Based on Mechanism of Action
Derived from their mechanism of action, antiviral drugs can be classified into distinct categories including Nucleoside Reverse Transcriptase Inhibitors (NRTIs), Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs), Protease Inhibitors (PIs), CCR5 Antagonists, Fusion Inhibitors, and Neuraminidase Inhibitors. Each class exhibits specific actions aligning with various stages of the viral life cycle, as depicted in Figure 3.
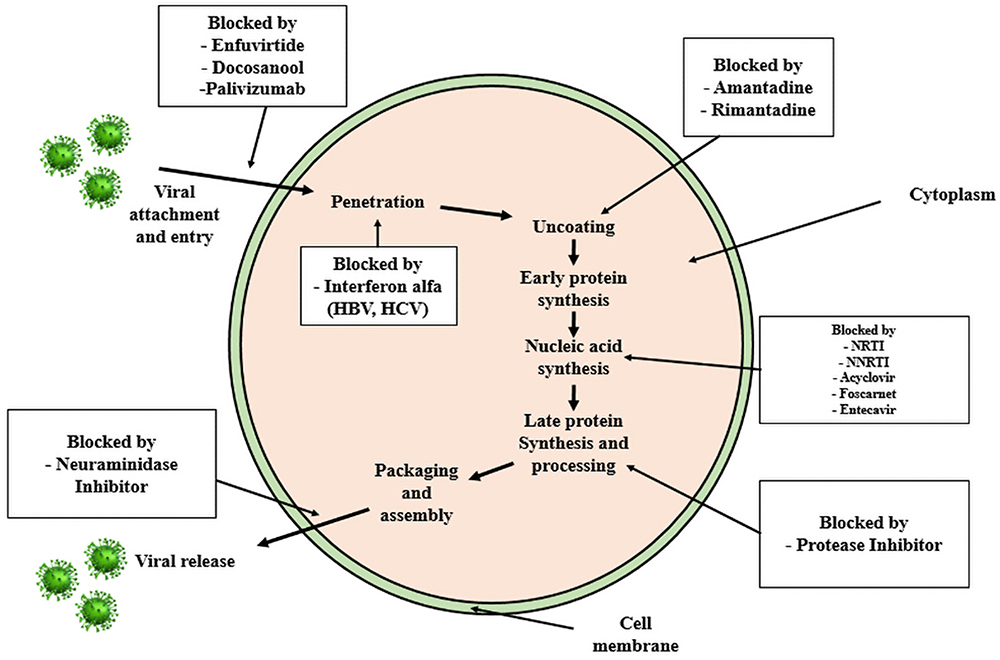 |
Figure 3 Antiviral Mechanism of Action. |
Nucleoside Reverse Transcriptase Inhibitors (NRTIs)
The NRTIs is still a vital part for the majority of combination regimens because it was the first class of antiretroviral medications to be approved for use in the United States. The HIV reverse transcriptase is inhibited by integrating into the nucleotide analogue, which results in DNA chain termination, or by competing with the virus’s natural substrate. The NRTIs are phosphorylated intracellularly to their active diphosphate or triphosphate metabolites.37
Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs)
In contrast to NRTIs, non-nucleoside reverse transcriptase inhibitors (NNRTIs) do not require intracellular phosphorylation to exert their pharmacologic effects. NNRTIs are noncompetitive inhibitors of reverse transcriptase, which alters the enzyme’s structure and reduces its activity.36
Protease Inhibitors (PIs)
Late in the HIV replication cycle, protease inhibitors (PIs) work pharmacologically by binding to HIV proteases and blocking the enzyme’s proteolytic activity, which prevents the development of mature and contagious virions. Certain ritonavir-boosted PI regimens are regarded as optimal therapy in patients who have never received treatment when combined with 2-NRTIs. Patients who experience treatment failure can also use PI plus NRTI combination regimens in conjunction.
CCR5 Antagonist
The only CCR5 antagonist that has been authorized for use in either treatment-experienced or treatment-naive individuals with CCR5-using viruses is maraviroc. The interaction between the HIV gp120 and the CCR5 receptor for CCR5-tropic HIV is blocked by its specific binding to the human CCR5 receptor on the cell membrane. It does not, however, prevent the entry of CXCR4-tropic HIV or HIV that enters cells via both CCR5 and CXCR4. Viral tropism testing must be carried out before maraviroc is prescribed to ensure that the patient’s virus only utilizes the CCR5 co-receptor.32
Fusion Inhibitor
Enfuvirtide, a fusion inhibitor, prevents the conformational changes required for the fusing of the viral and cellular membranes by binding to the first heptad-repeat (HR1) in the viral envelope glycoprotein gp41.63
Neuraminidase Inhibitor
Neuraminidase inhibitors (NAIs) are a type of medication that inhibits the neuraminidase enzyme. They are a type of antiviral medication that is frequently used to treat influenza. The ability of viral neuraminidases to facilitate viral budding from the host cell is crucial for influenza proliferation. This class includes the drugs oseltamivir (Tamiflu), zanamivir (Relenza), laninamivir (Inavir), and peramivir. NAIs act against both influenza A and influenza B, in contrast to M2 inhibitors, which only work against the influenza A virus. For the treatment and prevention of influenza A and B, the NAIs oseltamivir and zanamivir have received approval in the US and Europe. Peramivir inhibits neuraminidase activation for a lot longer than oseltamivir or zanamivir by tightly attaching to the neuraminidase of the influenza viruses. However, the delayed release of laninamivir from the cells into the respiratory system leads to long-lasting anti-influenza virus activity. As a result, laninamivir’s long-lasting action mechanism differs significantly from peramivir’s.64–67
Primary Consideration in the Development of Antiviral Drug Delivery System
In general, there are two main factors that requires modification of antiviral drugs in the form of nanoparticle delivery systems. First is the physicochemical issue, involving solubility and permeability. Poor solubility and permeability of active substance can be the primary cause of its poor bioavailability, which prevents the substance from exerting its full therapeutic potential. Addition of hydrophilic polymers in the creation of nanoparticle carriers and particle size reduction has been successful in improving the physicochemical issues of drugs related to it solubility and dissolution. Further, nanoparticles such as nanoliposomes are quite promising to solve the issue of a drug’s poor permeability due to its lipidic nature mimicking the cell membrane.
The second factor relates to the selectivity of the drug to the target site, which should be carefully considered for drugs with specific therapeutic targets such as certain genes, receptors, aberrant cells, or microorganisms. For instance, in the case of SARS-CoV-2, the knowledge that ACE 2 receptor serves as the infection target of the disease can be used as a basis for the modification or development of antiviral drugs that can competitively inhibit the receptor. Additionally, nanoparticles with active targeting can also be created by coating the nanoparticles with targeting ligands.68–70
Current Development of Antiviral Drugs
Nanoparticle Drug Delivery System for Antiviral Drugs
Nanomedicine is a major element of clinical therapy and is an application of nanotechnology. Nanocompounds have a large surface area due to their nanosize (1–100 nm), which improves surface contact with the solvent and accelerates the rate of dissolution of less water-soluble compounds.71 Therapeutic nanomedicine interventions have the potential to be incredibly precise at the intermolecular level, allowing for the treatment of diseases or the repair of damaged tissues like bones, muscles, or nerves. Liposomes, dendrimers, solid lipid nanoparticles, polymeric nanoparticles, silicon or carbon materials, metal, and magnetic nanoparticles are a few types of nanocarriers that have been created as drug delivery systems (Figure 4).72 A drug delivery system based on nanoparticles that integrates physics and chemistry disciplines is one potential approach of modification. It offers a targeted drug delivery system and is a tried-and-true effective solution to the issue of drugs with limited water solubility.73 In the last five years, modifications of nanoparticles for antiviral drugs have shown significant achievements in terms of their physicochemical properties and efficacy for inhibiting or preventing viral infections. Figure 4 shows the basic construction of the nanoparticle system.
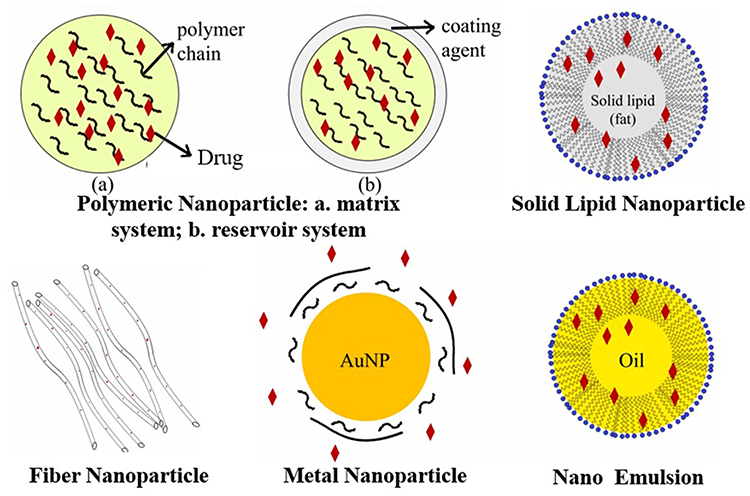 |
Figure 4 Nanoparticle Drug Delivery System: a. Matrix System of Polymeric Nanoparticle; b. Reservoir System of Polymeric Nanoparticle. |
The combination complex (nano-conjugates) of two FDA-approved medications, sitagliptin (SIT) and glatiramer acetate (GA), was to be tested in 2021 against a human isolate of the SARS-CoV-2 virus. In accordance with a full three-factor bilevel (23) factorial design, SIT-GA nano-conjugates were created. The factors used were the SIT concentration (mM, X1), GA concentration (mM, X2), and pH (X3). Zeta potential (mV, Y2) and particle size (nm, Y1) were evaluated as responses. The analysis of the data showed that the synthesized complex’s optimum formula was a 1:1 SIT:GA molar ratio at a pH of 10, which satisfied the necessary requirements with a desirability value of 0.878 and had particle sizes and zeta potentials of 77.42 nm and 27.67 V, respectively. The SIT-GA nano-complex demonstrated antiviral activity against a SARS-CoV-2 isolate with IC50 values for SIT, GA, and SIT-GA nano-conjugates of 16.14, 14.09, and 8.52 µM, respectively. The formula’s components have a high affinity for the COVID 3CL protease, which is necessary for coronavirus replication and may be inhibited (IC50 = 2.87µ M) by molecular docking. Both higher cellular absorption and enhanced delivery to target cells could be ensured by optimizing SIT-GA’s formulation.45
Ag/ZnO composite colloidal nanoparticles were created by Mahboubeh et al in 2022, and the surface of the nanoparticles was enhanced by an amodiaquine ligand. The results show that a synthetic colloid with a concentration of 15 g/L (Ag)/50 g/mL (ZnO) removed more than 7 logs of bacteria, fungus, and viruses. This eradication for the Covid 19 virus takes 30 seconds to go from 3.2 x 108 numbers to 21 viruses. Additionally, testing on the synthetic colloid’s toxicity and irritation reveal no harmful effects on human cells or tissues. 500 persons who had been exposed to the coronavirus participated in clinical trials using these colloidal nanoparticles as a mouthwash solution. According to the findings, depending on the severity of the condition, each patient’s recovery from illness occurred at a different time after washing their mouth and nose three times a day. During the trial, the virus did not infect nearly all of the participants who had no symptoms of infection and were using this solution as mouthwash.47
Adeola et al conducted a different study in 2022. They assessed the efficacy of nano-monocaprin (NMC) in inhibiting both phi6 and MS2 bacteriophages within an encapsulated environment. Using the sonochemistry method, NMC was created. Dynamic light scattering measurements of the formed 8.40.2-nm NMC were confirmed by size and shape analyses by contrasting the antiviral activity of NMC and molecular monocaprin (MMC) against phi6, which was employed as a stand-in for SARS-CoV-2, at doses of 0.5 mM and 2 mM. Using a plaque assay, the synthesized NMC shown 50% more antiviral activity against phi6 than MMC at pH 7. At pH 4, NMC more effectively inactivated phi6 than at pH 7. Authors employed the MTS assay to determine NMC’s IC50 for the HPDE and HeLa cell lines, which were 203 and 221 µM, respectively, to determine whether it is hazardous to mammalian cells. Since many viruses enter the human body through the mucosal lining of the nose, eyes, and lungs, NMC may be applied as a prophylactic drop or spray.15
By simply directing the development of interspatial 3D networks of hydrogels through a flawless locking mechanism, Biswajit et al in 2022 conducted in situ nanostructured and polysaccharide encapsulated ZnO NPs are generated with high antiviral effectiveness and low cytotoxicity (%viability 90%). Cell viability for the two composites, ChH@ZnO and ChB@ZnO, is 93.6% and 92.4%, respectively, up to a concentration of 400 μg/mL for the human cytomegalovirus (HCMV). This study offers important new information about the impact of ZnO NPs surface coatings on their nanotoxicity and antiviral activity, and it may help in the creation of more effective antiviral medications.43
Anthony et al carried out another investigation in this field (2022). The study shows that a functionalized nano-graphene oxide coating made from coal may be applied to fabrics and retain its antiviral capabilities even after being mechanically abraded or bleached. Chemical exfoliation of low-cost coal yields nano-graphene oxide, which is then functionalized with octadecylamine to produce repellent characteristics. In order to improve coating adherence and liquid repellency, functionalized nano-graphene oxide is applied to polyethylene terephthalate (PET) fabric following wet etching. The endurance and repellency are further increased by adding a second layer of polydimethylsiloxane (PDMS) on top of the functionalized nano-graphene oxide. The PDMS/functionalized nano-graphene oxide covering effectively repels saliva and water droplets. Furthermore, even after mechanical abrasion and bleach washing, we show that human adenovirus type 5 (HAdV5), herpes simplex virus type 1 (HSV-1), and betacoronavirus (CoV) have antiviral characteristics. The coating lowers the titers of CoV, HSV-1, and HAdV5 by 2.4 log (99.6%), 2.2 log (99.4%), and 1.8 log (98.6%), respectively. The coating may be used in large-area, high-production coating applications or reusable, antiviral personal protection equipment.44
Current findings on nanoparticles for antiviral drugs was conducted by Mohamed et al, 2023. PEGylated bilosomes (PBs), which contain PEGylated edge activator in addition to the standard components (Span 60, cholesterol, and bile salts), were proposed to increase RSV’s permeability and bioavailability. The design of 23 factorial experiments was used to investigate the significant influence of various variables on the features of the vesicles and select the optimal formula. Particle size (PS), zeta potential, and entrapment efficiency percent (EE%) were used to evaluate the formulas (ZP). The ideal formula (F5), which has a spherical shape, also has EE% of 86.12.9%, PS of 228.98.5nm, and ZP of 39.81.3mV. The superior dissolving behaviors of the sorted optimal formula (F5) were observed, and it increased Caco-2 cells’ cellular absorption by approximately 4.7 folds in comparison to RSV dispersion. Additionally, F5 showed 6.6 times greater antiviral activity compared to RSV dispersion and full in vitro suppression of SARS-CoV-2 at a dose of 0.48 g/mL. Resveratrol and the important residues of the SARS-CoV2 Mpro enzyme may interact, as demonstrated by the successful molecular modeling. As a promising oral panel of RSV for the treatment of SARS-CoV-2 infection, F5 may be suggested.46
The studies conducted by Rojekar et al in 2021 and 2022 focus on the development and evaluation of selenium nanoparticle-modified formulations for improving anti-HIV therapy. In the 2021 study, nanostructured lipid carriers of Etravirine were developed and modified with nano-selenium using a double emulsion solvent evaporation method. The dual-loaded nanocarrier system demonstrated higher efficacy against HIV1-infected cells compared to the plain drug, attributed to the synergistic effect of nano-selenium. Confocal microscopy and flow cytometry results showed enhanced uptake in cells, and in vivo studies indicated a significant increase in antioxidant levels, suggesting potential protective effects. The dual-loaded formulation also exhibited improved pharmacokinetic parameters and higher accumulation in remote HIV reservoir organs. This study suggests the potential of a dual-loaded formulation for synergistically targeting HIV1 infection while improving intracellular antioxidant balance for prolonged anti-HIV therapy.8
In the 2022 study, mannosylated lipid-based carriers were developed to deliver Etravirine and Selenium nanoparticles to HIV reservoirs via the mannose receptor. The nanocarrier system demonstrated higher effectiveness and a significant increase in the therapeutic index compared to the plain drug, as observed in in vitro anti-HIV1 efficacy assessments using infected cells. Confocal microscopy and flow cytometry studies indicated efficient uptake of the formulations. The study also demonstrated the protective effect of nano-selenium against oxidative stress in rats, highlighting its potential in mitigating oxidative stress induced by long-term therapies. In vivo biodistribution assessments showed significant improvements in brain, ovary, and lymph node biodistribution compared to the plain drug. The combination of delivering drugs via mannosylated nanostructured lipid carriers, coupled with the inclusion of Nano-Selenium, emerges as a promising and efficient approach for drug delivery to HIV reservoirs while concurrently addressing oxidative stress induced by prolonged therapies.9
In 2021, Faheem et al also conducted studies about anti-viral drug structural modification. Chitosan (CS) and Starch (SR), which act as hydrophobic drug carriers, were copolymerized as biodegradable nanohydrogels and functionalized with phthalic anhydride and hexamethylenetetramine, respectively, using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide catalyzed coupling. As a result of hydrophilic interactions with aqueous solutions, key features like porosity rise and crosslinking density decrease with consecutive functionalization. The hydrogel matrices with uniform particle size, porosity, and deep pores with high internal surface area were found by FESEM analysis to be capable of interacting with the drug and biomolecules to produce effective drug delivery. Hydrophobic-anionic model drug (Bromocresol green) release and the impact of induced functionalities were investigated under physiological conditions. The manufactured nanohydrogel’s capacity for drug release was successively improved from 65% to 80% and 85%. Due to stronger H-bonding and entanglement within the system, which was precisely controlled by the induction of hydrophilic, flexible, and biocompatible characteristics in term of expanded interfaces for the drug solutions, the drug administration in selective hydrogel was not relevant. The selective hydrogel was proposed as promising carriers for the hydrophobic anionic medicines under physiological conditions based on physicochemical and electrokinetic results.48
Liposome Formulation for Antiviral Drug
Liposome nanoparticles (nanoliposomes) are liposomes with particle sizes ranging from 80 to 300 nm. Due to their ability to distribute pharmaceuticals, liposome nanoparticles can enhance the physicochemical characteristics and performance of drugs. Using the thin-film hydration approach with -mangostin as a pharmacological payload, Chen created a liposome. The blood-brain barrier (BBB) was crossed at a rate of about 210 /cm2 with a horseradish peroxidase (HRP) permeability of less than 5% using the intercellular distribution experiment, which revealed a time-dependent feature for liposomes.74 The structural layout of the liposome is visible within Figure 5.
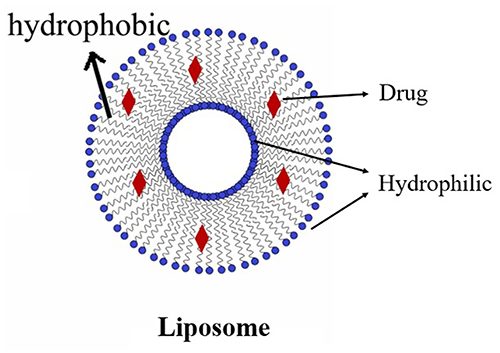 |
Figure 5 Liposome. |
Viveca et al developed a liposome formulation for an antiviral medication in 2021. Here, a number of 2-aminomethyl-3-hydroxy-1,4-naphthoquinone derivatives with n-butyl, benzyl, and nitrobenzene substituents in the main amine of naphthoquinone were loaded into liposomes. They were found to have strong HSV-1 inhibitory activity in the past. The early and late phases of HSV-1 replication could be stopped by any of the aminomethyl-naphthoquinones derivatives encapsulated in phosphatidylcholine liposomes, but those substituted with benzyl (compound 2) and nitrobenzene (compound 3), which produces selective in-dex values that are nearly nine times more effective than acyclovir, were especially effective. Their decision to use liposome as a medication carrier of aminomethylnaphthoquinones derivatives for formulations of in vivo pre-clinical testing is supported by the industry’s increased interest in topical treatment against HSV.17
Ilina et al conducted out another investigation in 2022 regarding liposome formulation for antiviral drug. The study has used vesicles made from plasma membranes to control a viral infection. We demonstrate the prophylactic and therapeutic efficacy of plasma membrane-derived liposomes in the treatment and prevention of herpes simplex virus type 1 (HSV-1) infection. In this investigation, human corneal epithelial (HCE) cells, which are naturally susceptible to HSV-1 infection, as well as Vero and Chinese hamster ovary (CHO) cells’ plasma membrane liposomes were employed. In contrast to CHO-derived liposomes, which lack the necessary HSV-1 entrance receptors, the study conclusively shows that HCE and Vero-derived cellular liposomes, which express the viral entry-specific cell surface protein receptors, exhibit robust antiviral action. Similar outcomes were obtained from additional testing of the plasma membrane-derived liposomes with HSV type-2 (HSV-2) and the pseudorabies virus, suggesting a high potential for using these liposomes to explore viral entrance mechanisms in a cell-free environment.49
In order to increase piperine’s water solubility and subsequently its therapeutic effectiveness, Mohamed et al enclose it inside bile salt-based nano vesicles in 2021. To study the effects of various formulation variables on the characteristics of bilosomes, piperine-loaded bilosomes were created using thin film hydration technology in accordance with a 32.21 full factorial design. These variables included entrapment efficiency (EE%), particle size, and percentage of drug released after 8 hours (Q8hr). The chosen best formula was F2, which had 1% bile salt, brij 72 as a surfactant, and a 9:1 ratio of surfactant to cholesterol. It displayed high EE% (97.2±0.8%) nanosized spherical vesicles (220.2±20.5 nm), as well as Q8hr (88.2%±5.6). Ex vivo permeation studies and pharmacokinetic studies both demonstrated the advantages of the optimized formula (F2) over the drug solution and the increased oral bioavailability of piperine-loaded bilosomes in comparison to piperine suspension. Additionally, F2 had much higher anti-viral efficacy and a higher safety margin than the medicine that was suspended. The docking structure of piperine, which shows that it can interact with the important amino acids in the receptor binding domain 4L3N, explained its reported activity. Additionally, in MERS-CoV-infected mice, F2 drastically lowers oxidant indicators and inflammatory cytokines. As a result, piperine may be best transported via bilosomes, which also have potential antiviral and anti-inflammatory effects.56
Cocrystal Modification for Antiviral Drugs
A co-crystal is a multicomponent crystal that comprises noncovalent interactions including hydrogen, van der Waals, and ionic bonds within its crystal lattice, all of which are normally solid at ambient temperature in a stoichiometric ratio.75 Co-crystals in a crystal lattice contain pharmaceutically approved guest molecules in addition to the API. Co-crystallization, which results in co-crystals, can improve the physiochemical properties of pharmaceuticals. Co-crystallization with pharmaceutically acceptable (GRAS) compounds had no effect on the pharmacological efficacy of the API, but it could enhance its physical qualities, such as its solubility, hygroscopicity, and compaction behavior.76 Pharmaceutical co-crystals are solids at room temperature that consist of an API and one or more distinctive co-crystal formers. Pharmaceutical co-crystals benefit from crystal engineering’s ability to improve physical and chemical properties. A pharmaceutical co-crystal is produced by combining an API and a co-crystal former into a single crystalline solid, according to earlier research.77 Several interactions, including hydrogen bonding, π–π-stacking, and the van der Waals forces result in the formation of co-crystals. Although by this definition API solvates and hydrates are not considered co-crystals, the crystal lattice of co-crystals may contain one or more solvent or water molecules. By altering the solubility, dissolving rate, mechanical behavior, moisture uptake, physical and chemical stability, and bioavailability of nonionizable medicines without altering their pharmacological activity, co-crystals enhance medicinal qualities.78,79 The visual representation of the co-crystal is depicted in Figure 6.
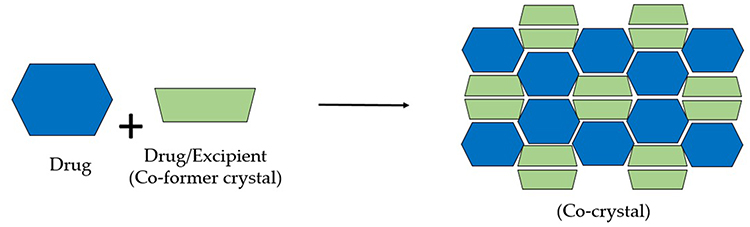 |
Figure 6 Cocrystal Modification. |
Ling et al conducted study in 2020 As the first example of a dual-drug cocrystal concurrently combining an antiviral medicine and an antibacterial agent, amantadine hydrochloride (AMNH) and sulfathiazole (SUZ) are conceived and successfully manufactured. A detailed description of the SUZAMNH cocrystal’s structure is provided. According to single crystal X-ray diffraction, the cocrystal structure contains a 1:1 ratio of the two components, and the interactions between the molecules of SUZ and AMNH are primarily dominated by N-H-O hydrogen bonds and N-H+ (AMNH)-Cl-H-N(SUZ) charge-assisted hydrogen bonds, creating a three-dimensional supramolecular structure. Under physiological pH conditions, the cocrystal’s dissolubility and permeability are extensively investigated. In comparison to pure SUZ, it has been discovered that SUZ in the cocrystal exhibits an increase in water solubility of 1.83–5.23 times and an improvement in penetrability of about 2-fold. The enhancement of antibacterial activity for SUZ might interestingly be attributed to simultaneously improved physicochemical features, showing enhanced inhibition of bacterial strains and lower values of minimum inhibitory concentration. The current study not only offers a potential method for therapeutic hybridization of antiviral and antibacterial medications against coinfection, but it also uses a novel pathway of the drug-drug cocrystallization strategy to expand the range of new applications for traditional medications.18
Ling et al establish a cocrystallization strategy for bidirectional coupling optimization in 2021, the following year. This method uses ATHC’s high water solubility to increase RVA’s dissolubility and bioavailability, so activating its auxiliary antiviral activity, while suitably reducing ATHC’s excessive solubility. To provide a synergistic impact for the cocrystal, the per-fected antiviral activity of RVA cooperates with ATHC in antiviral efficacy. The first antiviral drug–nutritional crystal, ATHC-RVA, has been produced and structurally characterized using this method. The solubility and bioavailability of RVA have improved by 152 and 9.64 times, respectively, according to in vitro/in vivo property evaluations carried out using theoretical and experimental methods. The risk of toxic effects brought on by the excessive solubility of ATHC is also decreased through a slower release. Particularly, CI 1 exhibits a potential to overcome medication resistance by greatly increasing the antiviral synergy of ATHC and RVA. All of these advantages create a unique route for the cocrystallization of synergistic antiviral pharmaceutical cocrystals.55
Recently, Dattatraya et al (2023) attempted to use a pharmaceutical cocrystallization approach to increase the oral bioavailability of the antiviral medication Efavirenz (EFV). Due to its exceedingly poor water solubility and low oral bioavailability, EFV falls under BCS-II. Efavirenz nicotinamide cocrystal (ENCOC) was made using the liquid-assisted grinding technique after EFV and nicotinamide (NICO) were chosen in a (1:1) stoichiometric ratio (LAG). Spectroscopic methods such as Fourier transmission infrared (FTIR), Raman, and 13C solid-state nuclear magnetic resonance were used to confirm the creation of a new solid phase (13C ssNMR). The thermal behavior and melting patterns of ENCOC, EFV, and NICO were seen using thermal techniques such as differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), and hot stage microscopy (HSM). It is confirmed by X-ray powder diffraction (XRPD) that ENCOC has formed a new crystalline phase. Through the use of scanning electron microscopy, the morphology was assessed (FESEM). Studies on saturated solubility and in vitro drug release showed improvements in solubility of 8.9 times and percentage cumulative drug release of 2.56 times, respectively. More than 97% of the medication was discovered to be present in ENCOC, and the crystal displays exceptional accelerated stability. In comparison to EFV with Efcure®-200 tablet (2896.21 ng/mL), the oral bioavailability of EFV (Cmax, 799.08 ng/mL) shows a significant improvement after cocrystallization (Cmax, 5597.09 ng/mL). The current study presents a scalable and economical strategy for improving the solubility, oral bioavailability, and physicochemical stability of an antiviral drug called EFV.
Solid Dispersion for Antiviral Drugs
Solid dispersion is a formulation strategy used in pharmaceuticals to improve the solubility and bioavailability of poorly water-soluble drugs (Figure 7). This approach involves dispersing the drug in a solid matrix, often a polymer, to enhance its dissolution properties. While solid dispersion has been predominantly explored for improving the solubility of various drugs, including antiviral drugs, its application in the context of antiviral medications is not as widespread as in some other therapeutic areas. However, researchers continue to explore innovative drug delivery approaches to improve the performance of antiviral drugs, and solid dispersion is one such avenue.
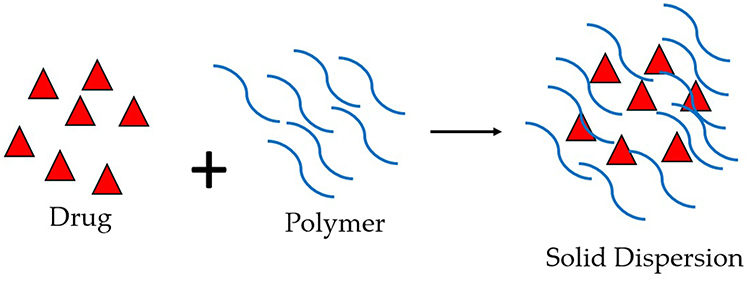 |
Figure 7 Solid Dispersion. |
The three referenced studies on solid dispersion for antiviral drugs contribute valuable insights to the scientific literature, each employing distinct methodologies and focusing on different aspects of drug formulation. In Mazumder et al’s work (2017), polysaccharide-based polymers, specifically cellulose acetate-based, were utilized to produce nanoparticles via a rapid precipitation process. Their study delves into the structure-property relationships of these polymers, emphasizing the solubility enhancement of poorly soluble drugs. The nanoparticles exhibited increased solubility and faster drug release compared to pure drugs, with the formulation process and nanoparticle properties influencing drug solubility.13
Damien F. et al (2002) investigated the physical stability of solid dispersions of UC-781 with various carriers, including PEG 6000, Gelucire 44/14, and PVP K30. Their study explored the impact of storage conditions on physicochemical properties using DSC, X-ray powder diffraction, and dissolution studies. The observed changes in carrier properties over time were linked to decreased dissolution properties, highlighting the importance of carrier stability in solid dispersions.53
In Damien F. et al’s earlier work (2000), solid dispersions of the antiviral thiocarboxanilide UC-781 with PEG 6000 and Gelucire 44/14 were prepared using the fusion method. Their study focused on the characterization of dispersions through dissolution studies, DSC, FTIR, and X-ray powder diffraction. Notably, the dissolution of UC-781 showed a significant improvement in solid dispersions compared to its pure form, and the absence of well-defined drug-polymer interactions was established.54
Salt Modification for Antiviral Drugs
The solubility enhancement strategy involving the synthesis of less water-soluble salts on the active ingredient employs the principle of chemical structure modification as a means to augment solubility. According to the findings, the active component will dissolve far more quickly in its salt form than in its original state. This is due to how quickly the salt ionizes in the solvent media. Usually, bioactive compounds that are not very soluble in water are the ones that are modified with salt approaches. Salt modification can make an active molecule more soluble, although in some cases the resulting salt is less stable Although adding salt to a medicine makes it more soluble in water, the technique needs to be adjusted for the desired dissolution medium.80–82 The visual representation of salt modification is evident in Figure 8.
 |
Figure 8 Salt Modification. |
In 2020, Ling et al devised and successfully synthesized the first molecular salt combining the antibacterial medication sulfathiazole (SFZ) and the antiviral component amantadine (ATD) using a salification technique. By using single-crystal X-ray diffraction and other methods, the exact structure of the recently discovered dual drug molecular salt SFZ-ATD has been solved. The single-crystal diffraction analysis confirms that the proton transference from SFZ to ATD molecules results in the composition for the molecular salt with an equal amount of SFZ and ATD, where the supramolecular network in the crystal is primarily controlled by charge-assisted one-dimensional hydrogen-bonding chains and two-dimensional hydrophobic layer. Both theoretical and experimental studies are conducted on the pertinent molecular salt characteristics. The experimental results show that SFZ in the molecular salt exhibits improvements in both permeability and dissolubility when compared to pure SFZ. This is further corroborated by the theoretical calculation of molecular electrostatic potential. What’s more, the benefits of improved physicochemical profiles for the dual-drug salt result in SFZ’s enhanced antibacterial activity against the studied bacterial strains. Thus, this work not only gives a chance for the regeneration of traditional sulfa medicines but also some fresh concepts for the salinization-based therapeutic hybridization of bacteria and viruses.14
Sulfamethoxazole (SFM), an antibacterial medicine, and amantadine (ATE), an antiviral component, were effectively combined to create a novel dual-drug crystalline molecular salt called SFM-ATE in 2021. This salt was created using a multidrug salification technique guided by proton exchange reaction. Single-crystal X-ray diffraction and a number of other techniques are used to precisely identify the crystal structure of the first molecular salt that was formed. The findings demonstrate that the traditional hydrogen bonds and charge-assisted hydrogen bonds lead to the formation of two-dimensional networks in the crystal lattice of the molecular salt SFM-ATE, where the hydrophobic interaction is significant. The dual-drug molecular salt is investigated for its pertinent in vitro/vivo medicinal properties using both theoretical and experimental approaches in comparison. After generating the molecular salt, it has been discovered that SFM exhibits concurrent gains in permeability and dissolution over the bulk medication. This finding is substantiated by calculations of the molecular electrostatic potential and Hirshfeld surface analyses. It is reassuring to see that the idealized in vitro biopharmaceutical features can successfully translate into the in vivo pharmacokinetic preponderances with the accelerated peak plasma concentration, prolonged half-life, and improved bioavailability. Even better, as inhibition regions grow and the values of minimum inhibitory concentrations against the studied bacterial strains decrease, SFM from the molecular salt exhibits higher antibacterial properties. Therefore, the current contribution offers an alternative strategy for treating viral-bacterial coinfection and other complex diseases by drug hybridization at the molecular level, in addition to providing an opportunity for expanding applications for traditional sulfa drugs via dual-drug salification strategy.55
Nikolai et al, in 2021. Proposed practical procedures for the synthesis of pyrazoline N-alkylidene salts based on terpene (camphor, camphorquinone, carvone) ketones, cage (adamantanone and norcamphor) ketones, and natural aldehydes (carvone and myrtenal), permitting the isolation of stable pyrazolinium salts in individual form. The conditions for the synthesis of the intended products were optimized. One of the investigated compounds, 1-bornylidene-3-phenylpyrazolinium tetrafluoroborate, had the strongest antiviral effects against the influenza A/Puerto Rico/8/34 (H1N1) virus (IC50 = 6.2 μM, SI 107) of the tested compounds.58
In a different study from Jinsoo et al, in 2022, ZnO nanorods and different copper salts were built into Polyacrylonitrile (PAN) nanofibers that were electrospun to create antiviral activity in response to visible light. Using HR-TEM, the morphologies of ZnO nanorods were studied, and optical characteristics were assessed using UV-vis spectroscopy. The antiviral activities of the created Cu-ZnO/PAN nanofibers were assessed using ux174 under visible light irradiation, and the electrospun nanofibers were described using SEM and X-ray spectroscopy. Due to ZnO nanorods’ ability to operate as a photocatalyst, the CuBr and ZnO nanorod-containing nanofibers displayed exceptional antiviral activity.56
Shuang et al, in 2022 also required the creation of an enabling formulation for human clinical investigations in order to attain the requisite therapeutic in vivo concentration of dasabuvir. Although salt creation has been used frequently to improve the solubility and dissolution rate of solids, it has seldom been employed to create oral solid dosage forms for acidic medications as weak as dasabuvir due to worries about the free acid’s quick disproportionation and crystallization. The study delineates endeavors in this contribution to identify dasabuvir monosodium monohydrate as a pharmaceutical substance characterized by stability, manufacturability, and, notably, substantial enhancements in the dissolution and oral absorption profiles of this inadequately soluble therapeutic agent. The triple-combination direct-acting antiviral HCV regimen, Viekira Pak, has been commercialized thanks to the oral dasabuvir administration made possible by the salt method. Other insoluble compounds can benefit from the methodologies and solutions found in targeted studies to address the technical difficulties encountered along the way (eg, incorporation of polymers to prevent crystallization and disproportion-ation and species mapping to enable salt manufacturing process).57
Structure Modification for Antiviral Drugs
The creation of APIs usually employs structural alteration to enhance solubility, dissolution, and bioavailability (Figure 9). Since the ultimate goal of structural alteration is the creation of a novel medicine, pharmacological activity and drug stability are two important factors to take into account throughout research and innovation. The physicochemical and biochemical characteristics are altered for pharmacokinetics and drug safety in order to improve the pharmacological activity and stability. The basic idea behind chemically altering the structure of active pharmaceutical compounds to increase solubility is to concentrate on adding or substituting hydrophilic groups, specifically carboxylate, amine, ester, ketone, and hydroxy, in the structure of new molecules. Otherwise, studying the pharmacophore and designing the structure by incorporating or removing both features hydrophilic or lipophilic is required in order to improve the activity of the drug.
 |
Figure 9 Structure Modification. |
In 2019, Belen et al synthesized a novel series of dendrimers with divalent and tetravalent branching arms in place of the prototype’s trivalent ones, and they evaluated their efficacy against HIV, EV71, a panel of 16 other viruses, and other diseases. These chemicals have been synthesized using divergent or convergent methods. Our results show that only compounds with tetravalent branched arms exhibited the same anti-HIV and anti-EV71 activity of the prototype (low micromolar) and even gained notable antiviral activity against new pathogens like HSV-2, adenovirus-2, human corona virus, and respiratory syncytial virus, being the first members of the Trp dendrimer family to exhibit activity against those viruses. These substances demonstrated low-nanomolar efficacy against a representative EV71 clinical isolate as the prototype did as well. The most potent IIa, which contains tetravalent branching arms, interacts with the viral envelopes of HIV, EV71, and HSV-2, which may impede virus attachment to the host cell, according to ongoing experimental research to discover its mode of action. These findings confirm the potential of this new class of Trp den-drimers and establish them as valuable prototypes for the design of novel viral entry inhibitors with wide antiviral activity.59
Kiril et al conducted a different study in 2019. The antiherpetic medications ganciclovir, penciclovir with the bile acids (choline, chenodeoxycholine, and deoxycholine), and amino acid esters of acyclovir were created and their in vitro antiviral activity against herpes simplex viruses type 1 and type 2 was assessed (HSV-1, HSV-2). The antiviral assays showed that modified analogs of ACV and PCV are less effective against HSV-1 and HSV-2 than the original drugs. Ganciclovir-CC50 deoxycholate’s matched those of the other analogs, and it exhibits lower activity than ganciclovir. The obtained results demonstrate that the modified nucleoside analogs’ bioavailability in cells is not improved by the tested change.60
Elena et al carried out some research-related structural change of an antiviral medication in 2021. Through a coupling reaction with the siloxanes 3-glycidoxypropyl-trimethoxysilane (GLYMO), 3-methacryloxypropyl-trimethoxysilane (MEMO), and 3-mercaptopropyl-trimethoxysilane, three distinct functionalities have been added to mesoporous materials (MPTMS). Different functional groups’ locations and the way they interact with the mesoporous substrate have been determined. In addition to the accessible surface area, the amount of the antiviral medication acyclovir (ACV) adsorbed depends on the chemical or physical interactions between functions. The GLYMO and MPTMS functionalized materials’ drug adsorption isotherms follow mechanisms that are based on the materials’ various surface coverages and the potential for establishing physicochemical interactions between the drug molecules and the functionalities. On the other hand, the primary adsorption process when functionalizing with MEMO is typical of chemically bound adsorbates. In all the functionalized materials, the Weibull model best describes the ACV release kinetics. Drug diffusion occurs along the mesoporous channels at low kinetics and uniformly when the MTPMS is utilized as a functionalizing agent.61
According to Zhen et al (2021), ebselen, a multipurpose medication candidate, has recently been discovered to be a Mpro inhibitor. In order to increase potency, antiviral activity, and selectivity, we docked ebselen to the binding pocket of the Mpro crystal structure. To verify this theory, we created and synthesized ebselen derivatives with the goal of strengthening Mpro’s non-covalent bonds. In HPLC and FRET experiments, ebselen derivatives (0.3 μM) were tested for their ability to inhibit Mpro. With an IC50 range of 0.07 to 0.38μ M, nine ebselen derivatives (EBs) showed a higher inhibitory impact on Mpro. Further analysis of three derivatives revealed that EB2-7, with an IC50 value of 4.08 µM in HPAepiC cells as opposed to the prototype ebselen at 24.61 µM, exhibited the most powerful inhibition of SARS-CoV-2 viral multiplication. In the LC-MS/MS test, EB2-7 works as a noncovalent Mpro inhibitor. Together, our discovery of ebselen compounds with enhanced antiviral activity may pave the way for the future development of drugs to treat COVID-19 and SARS-CoV-2 infection.62
Additional research by Hanieh et al, 2022: It has been acknowledged that in vitro transcribed (IVT)-mRNA is a promising therapeutic approach. IVT-mRNA is a desirable replacement for protein- or virus-based medications due to advancements in simple and quick production technologies. Its clinical application is aided by robust expression levels, a lack of genotoxicity, and controllable immunogenicity. We proposed that combinations of 50 -end and internal IVT-mRNA alterations may be used to tune or abolish the innate immune responses of therapeutically important human cells. Our results demonstrate the critical role of uridine alterations in IVT-mRNA performance using primary human macrophages as targets. Five nucleotide modification techniques were examined, and 5-methoxy-uridine performed better than the others, increasing transgene expression by up to fourfold while also inducing mild pro-inflammatory and undetectable antiviral reactions. Following HPLC purification, macrophage reactions to IVT-mRNAs with high immunogenicity (such as pseudouridine) may be reduced. On the other hand, the impact of 50 -end changes on mRNA expression and immunological responses was minimal. Our findings showed that human macrophages are affected by the absorption of chemically modified IVTmRNA, exhibiting unique innate immune response patterns along with increased temporary transgenic expression. Our research is expected to be useful in predicting the exact cell responses needed for a variety of therapeutic applications, such as generating regulated immunogenicity in mRNA vaccines and fully eliminating cell activation in protein replacement therapies.16
Molecular Pharmaceutics Perspectives on Mechanistic Pathways of Technique Modification
In the relentless battle against viral diseases, scientists wield a powerful arsenal of pharmaceutical techniques to optimize antiviral drugs. From the nanoscale to the molecular level, these strategies manipulate physicochemical properties and delivery mechanisms, ultimately boosting drug performance and paving the way for more effective treatments. Let us delve deeper into the intricate workings of each technique, exploring their unique contributions through the lens of molecular pharmaceutics:
Nanoparticle Drug Delivery Systems
Imagine tiny Trojan horses patrolling the bloodstream, loaded with antiviral ammunition. Nanoparticles, typically smaller than 100 nanometers, encapsulate drugs, shielding them from enzymatic degradation and facilitating targeted delivery to viral reservoirs. By exploiting specific physical or chemical interactions with viral components, nanoparticles can navigate complex biological barriers and release their cargo directly at the site of infection. This targeted approach not only maximizes drug efficacy at the infection site but also minimizes systemic exposure and potential side effects.
Liposomes
Picture microscopic bubbles composed of phospholipid bilayers, mimicking the cell membrane. Liposomes, these versatile vesicles, can encapsulate both hydrophilic and hydrophobic drugs, effectively overcoming solubility limitations. Upon encountering infected cells, liposomes fuse with the viral membrane, delivering their encapsulated antiviral payload directly into the viral replication machinery. This intracellular delivery bypasses cellular efflux pumps, a common mechanism of viral resistance, and potentiates drug action. For example, liposomal amphotericin B has shown increased efficacy against HIV compared to the conventional formulation, highlighting the power of liposomal delivery in tackling challenging viral infections.83
Co-Crystals and Solid Dispersions
Imagine drugs trapped in a molecular maze, their solubility enhanced by intimate contact with excipients. Co-crystals and solid dispersions tackle the hurdle of poor drug solubility by forming intricate molecular complexes with suitable excipients. In co-crystals, specific intermolecular interactions between the drug and excipient molecules lock them into a defined crystalline structure, improving dissolution and bioavailability. Solid dispersions, on the other hand, embed the drug within an amorphous matrix of the excipient, promoting rapid disintegration and drug release upon administration. Both approaches significantly increase the surface area of the drug available for absorption, leading to improved antiviral efficacy.84
Salt Formation
Envision a chemical handshake between a drug and an appropriate counterion, altering its charge and solubility. Salt formation involves converting a poorly soluble drug into its salt form, often by pairing it with an acidic or basic molecule. This ionic modification enhances aqueous solubility and dissolution rate, facilitating efficient drug absorption. Additionally, salt formation can alter the drug’s pKa, potentially influencing its distribution and metabolism.
Structure Modification
Imagine tweaking the molecular architecture of a drug, like tuning a musical instrument to resonate perfectly with its viral target. Structure modification involves carefully tailoring the chemical structure of the antiviral drug to optimize its interactions with viral proteins or enzymes. This can involve modifying functional groups, introducing steric bulk, or altering ring systems to achieve enhanced binding affinity and potency. Additionally, structure modification can be employed to reduce the susceptibility of the drug to viral mutations, thereby mitigating the emergence of resistance.
By harnessing these diverse pharmaceutical techniques, scientists can fine-tune antiviral drugs, transforming them into potent weapons against viral foes. From targeted delivery to enhanced solubility and potency, each approach offers unique advantages, paving the way for a future where more effective and targeted antiviral therapies become a reality.
Critical Evaluation
The critical characterization and evaluation of the mentioned modification techniques for improving antiviral drugs involve assessing several key parameters to substantiate their success. For nanoparticle drug delivery systems, comprehensive characterization should include size distribution, surface charge, drug loading efficiency, and in vitro release profiles. The success is often demonstrated through enhanced drug solubility, prolonged circulation time, and improved cellular uptake.
In the case of liposomes, critical evaluation involves determining encapsulation efficiency, stability, and membrane integrity. Successful liposomal formulations exhibit enhanced drug bioavailability, sustained release profiles, and improved therapeutic efficacy. Techniques such as dynamic light scattering and cryo-transmission electron microscopy can further validate the size, homogeneity, and morphology of liposomes.
For co-crystals, the confirmation of successful modification requires thorough characterization using techniques such as X-ray diffraction and differential scanning calorimetry to verify the crystalline structure and stability improvements. Evaluation also involves assessing dissolution profiles and pharmacokinetic parameters to ensure enhanced solubility translates to improved bioavailability.
Solid dispersions demand critical assessment through dissolution studies, differential scanning calorimetry, and powder X-ray diffraction to confirm amorphization and improved drug solubility. Successful solid dispersion formulations demonstrate superior dissolution rates and enhanced drug release compared to the crystalline form.
Salt formation success is typically confirmed through spectroscopic techniques like nuclear magnetic resonance and X-ray crystallography, ensuring the formation of stable salt forms with improved physicochemical properties. Evaluation extends to dissolution studies and bioavailability assessments.
Finally, structure modification success relies on elucidating the altered drug-receptor interactions and pharmacological effects. This involves sophisticated analytical techniques such as nuclear magnetic resonance, mass spectrometry, and molecular modeling to confirm structural changes and predict potential toxicity or improved antiviral activity.
In conclusion, the critical characterization and evaluation of these modification techniques involve a combination of physicochemical analyses, in vitro release studies, and in vivo assessments. The success of each technique is demonstrated by its ability to enhance drug solubility, stability, bioavailability, and overall therapeutic efficacy.
Conclusion
In general, the modifications made to antiviral drugs are based on the following issues: Firstly, the rise in the frequency and fatality rate of viral infections. When the coronavirus pandemic started in 2019, viral illnesses had already become one of the deadliest diseases in the previous three years. Secondly, antiviral medication is losing its effectiveness due to resistance brought on by viral mutations. Furthermore, the problem lies with the antiviral itself, specifically its physicochemistry, including solubility, selectivity, and stability.
Therefore, a number of modified strategies have been developed in an effort to address these issues. It has been discovered that some of the most often employed methods to solve the issue of antiviral medications over the past five years include salt alteration, structural modification, cocrystal, liposome, and nanoparticle drug delivery systems (Figure 10).
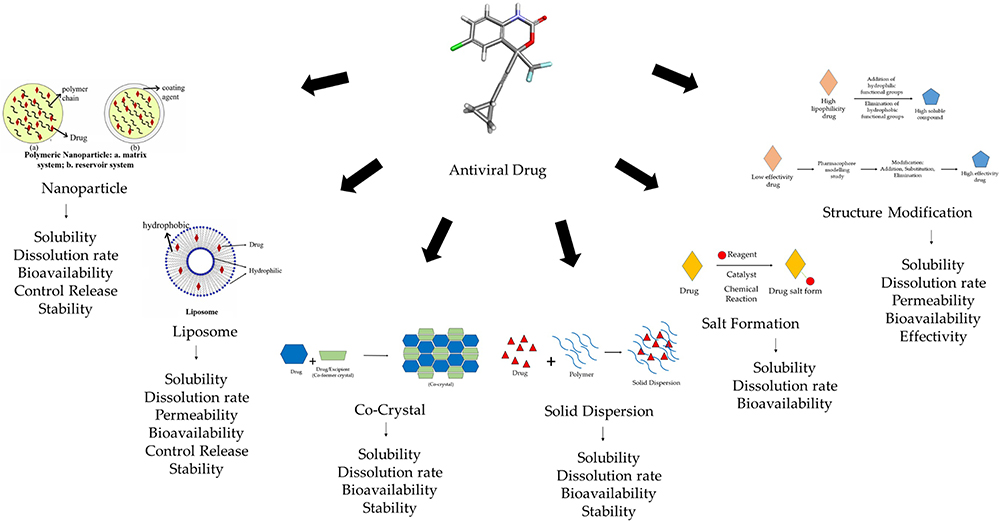 |
Figure 10 An Overview of The Modification Techniques Employed for Antiviral Drugs. |
Starting with the nanoparticle drug delivery system, which has the capability to accelerate wettability by increasing the surface area and contact area between the particles and the solvent medium while boosting solubility and dissolving rate. This is the approach’s main principle: by raising the concentration of high solutes and solubility rates, a large amount of drugs can be absorbed, enhancing bioavailability and pharmacological activity. Additionally, using a matrix or reservoir system that uses polymers as a release controller through swelling, erosion, and other mechanisms, nanoparticle technology is able to offer a customized release system. Currently, nanoparticles are also able to offer customized delivery systems, which may help a drug’s selectivity.85–88
The liposome drug delivery system comes next, which can greatly reduce the solubility and permeability issues of both hydrophilic and lipophilic drugs. The liposome’s two-layer structure, which comprises polar and nonpolar groups, can bind both hydrophilic and lipophilic substances. The essential concept of this technique is that the hydrophilic-hydrophobic bilayer has good partitioning in the solvent medium and physiological membranes of the body, which can linearly improve the bioavailability and pharmacological activity of medicinal molecules. Additionally, liposomes are effective at preserving the stability of therapeutic molecules based on proteins or genes.89–91
The next development, salt and cocrystal formation, aims to resolve a drug’s solubility issue. Cocrystals aim to combine medicinal compounds with crystal coformers to create crystals that are more soluble. A medication with weak basic properties will react with an acid, whereas a chemical with weak acidic properties will react with a base, and the end result of this reaction will generate a drug salt that is known to have high solubility. This method uses the acid-base principle, as opposed to salt creation.92–94
The modification of the structure comes next. In general, the objective of this modification strategy is to enhance the effectiveness and physicochemical characteristics. The study of the compound’s structure and pharmacophore emerges first. The results of this stage will inform us of the part of the structure or group that is in charge of producing a therapeutic effect, and conversely, we will gain knowledge about the part of the structure that is still subject to modification. For instance, we can change the structure of a medicine if it has physicochemical solubility issues by adding hydrophilic groups; alternatively, if it has permeability issues, we can consider including lipophilic groups.16,48,60,61
Overall, all alterations can offer spectacular benefits by addressing a drug’s physicochemical issues and enhancing its performance and efficacy.
Funding
This research was funded by Rector of Universitas Padjadjaran for article review grant scheme (1549/UN6.3.1/PT.00/2023).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Handayani D. Corona Virus Disease 2019. J Respirologi Indones. 2020;40(2). doi:10.36497/jri.v40i2.101
2. Sawinski D, Goral S. BK virus infection: an update on diagnosis and treatment. Nephrol Dial Transplant. 2015;30(2):209–217. doi:10.1093/ndt/gfu023
3. Napoli PE, Mangoni L, Gentile P, Braghiroli M, Fossarello M. A panel of broad-spectrum antivirals in topical ophthalmic medications from the drug repurposing approach during and after the coronavirus disease 2019 era. J Clin Med. 2020;9(8):2441. doi:10.3390/jcm9082441
4. Budiman A, Nurfadilah N, Muchtaridi M, Sriwidodo S, Aulifa D Lia, Rusdin A. The Impact of Water-Soluble Chitosan on the Inhibition of Crystal Nucleation of Alpha-Mangostin from Supersaturated Solutions. Polymers. 2022;14(20): 4370. doi:10.3390/polym14204370
5. Mahajan K, Rojekar S, Desai D, et al. Layer-by-layer assembled nanostructured lipid carriers for CD-44 receptor–based targeting in HIV-infected macrophages for efficient HIV-1 inhibition. AAPS Pharm Sci Tech. 2021;22(5):171. doi:10.1208/s12249-021-01981-4
6. Fotooh Abadi L, Damiri F, Zehravi M, et al. Novel nanotechnology-based approaches for targeting HIV reservoirs. Polymers. 2022;14(15):3090. doi:10.3390/polym14153090
7. Rojekar S, Abadi LF, Pai R, Mahajan K, Kulkarni S, Vavia PR. Multi-organ targeting of HIV-1 viral reservoirs with etravirine loaded nanostructured lipid carrier: an in-vivo proof of concept. Eur J Pharm Sci. 2021;164:105916. doi:10.1016/j.ejps.2021.105916
8. Rojekar S, Pai R, Abadi LF, et al. Dual loaded nanostructured lipid carrier of nano-selenium and Etravirine as a potential anti-HIV therapy. Int J Pharm. 2021;607:120986. doi:10.1016/j.ijpharm.2021.120986
9. Rojekar S, Abadi LF, Pai R, Prajapati MK, Kulkarni S, Vavia PR. Mannose-anchored nano-selenium loaded nanostructured lipid carriers of etravirine for delivery to HIV reservoirs. AAPS Pharm Sci Tech. 2022;23(7):230. doi:10.1208/s12249-022-02377-8
10. Abate C, Carnamucio F, Giuffrè O, Foti C. Metal-based compounds in antiviral therapy. Biomolecules. 2022;12(7):933. doi:10.3390/biom12070933
11. Žigrayová D, Mikušová V, Mikuš P. Advances in antiviral delivery systems and chitosan-based polymeric and nanoparticulate antivirals and antiviral carriers. Viruses. 2023;15(3):647. doi:10.3390/v15030647
12. Delshadi R, Bahrami A, McClements DJ, Moore MD, Williams L. Development of nanoparticle-delivery systems for antiviral agents: a review. J Control Release. 2021;331:30–44. doi:10.1016/j.jconrel.2021.01.017
13. Mazumder S, Dewangan AK, Pavurala N. Enhanced dissolution of poorly soluble antiviral drugs from nanoparticles of cellulose acetate based solid dispersion matrices. Asian J Pharm Sci. 2017;12(6):532–541. doi:10.1016/j.ajps.2017.07.002
14. Wang LY, Yu Y-M, Yu M-C, Li Y-T, Wu Z-Y, Yan C-W. A crystalline solid adduct of sulfathiazole-amantadine: the first dual-drug molecular salt containing both antiviral and antibacterial ingredients. CrystEngComm. 2020;22(22):3804–3813. doi:10.1039/d0ce00368a
15. Sorinolu AJ, Mamun MM, Vadarevu H, Vivero-Escoto JL, Vejerano EP, Munir M. Antiviral activity of nano-monocaprin against Phi6 as a surrogate for SARS-CoV-2. Int Microbiol. 2022;0123456789. doi:10.1007/s10123-022-00300-6
16. Moradian H, Roch T, Anthofer L, Lendlein A, Gossen M. Chemical modification of uridine modulates mRNA-mediated proinflammatory and antiviral response in primary human macrophages. Mol Ther Nucleic Acids. 2022;27:854–869. doi:10.1016/j.omtn.2022.01.004
17. Giongo V, Falanga A, De Melo CPP, et al. Antiviral potential of naphthoquinones derivatives encapsulated within liposomes. Molecules. 2021;26(21):6440. doi:10.3390/molecules26216440
18. Wang LY, Bu FZ, Li YT, Wu ZY, Yan CW. A Sulfathiazole-Amantadine Hydrochloride Cocrystal: the First Codrug Simultaneously Comprising Antiviral and Antibacterial Components. Cryst Growth Des. 2020;20(5):3236–3246. doi:10.1021/acs.cgd.0c00075
19. Lefkowitz EJ, Dempsey DM, Hendrickson RC, Orton RJ, Siddell SG, Smith DB. Virus taxonomy: the database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018;46(D1):D708–D717. doi:10.1093/nar/gkx932
20. Morgan GJ. What is a virus species? Radical pluralism in viral taxonomy. Stud Hist Philos Sci Part C Stud Hist Philos Biol Biomed Sci. 2016;59:64–70. doi:10.1016/j.shpsc.2016.02.009
21. Barr JN, Fearns R. Genetic instability of RNA viruses. In: Genome Stability. Elsevier; 2016:21–35.
22. Calisher CH. The taxonomy of viruses should include viruses. Arch. Virol. 2016;161(5):1419–1422. doi:10.1007/s00705-016-2779-x
23. Francki RIB, Fauquet CM, Knudson DL, Brown F. Classification and Nomenclature of Viruses: Fifth Report of the International Committee on Taxonomy of Viruses. Virology Division of the International Union of Microbiological Societies. Vol. 2. Springer Science & Business Media; 2012.
24. Morales-Sánchez A, Fuentes-Pananá EM. Human viruses and cancer. Viruses. 2014;6(10):4047–4079. doi:10.3390/v6104047
25. West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. 2017;17(6):363–375. doi:10.1038/nri.2017.21
26. Chin Y-W, Kinghorn AD. Structural Characterization, Biological Effects, and Synthetic Studies on Xanthones from Mangosteen (Garcinia mangostana), a Popular Botanical Dietary Supplement. Mini Rev Org Chem. 2008;5(4):355–364. doi:10.2174/157019308786242223
27. Domingo E. Virus as Populations: Composition, Complexity, Dynamics, and Biological Implications. Academic Press; 2015.
28. McLaughlin-Drubin ME, Munger K. Viruses associated with human cancer. Biochim Biophys Acta. 2008;1782(3):127–150.
29. Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63(6):1129–1136. doi:10.1016/0092-8674(90)90409-8
30. Cazalet C, Rusniok C, Brüggemann H, et al. Evidence in the Legionella pneumophila genome for exploitation of host cell functions and high genome plasticity. Nat Genet. 2004;36(11):1165–1173. doi:10.1038/ng1447
31. Lori F, Lisziewicz J, Smythe J, et al. Rapid protection against human immunodeficiency virus type 1 (HIV-1) replication mediated by high efficiency non-retroviral delivery of genes interfering with HIV-1 tat and gag. Gene Ther. 1994;1(1):27–31.
32. Baba M, Nishimura O, Kanzaki N, et al. A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc Natl Acad Sci. 1999;96(10):5698–5703. doi:10.1073/pnas.96.10.5698
33. Westby M, van der Ryst E. CCR5 antagonists: host-targeted antivirals for the treatment of HIV infection. Antivir Chem Chemother. 2005;16(6):339–354. doi:10.1177/095632020501600601
34. Leung D, Abbenante G, Fairlie DP. Protease inhibitors: current status and future prospects. J Med Chem. 2000;43(3):305–341. doi:10.1021/jm990412m
35. Nowak DA, Hermsdörfer J, Glasauer S, Philipp J, Meyer L, Mai N. The effects of digital anaesthesia on predictive grip force adjustments during vertical movements of a grasped object. Eur J Neurosci. 2001;14(4):756–762. doi:10.1046/j.0953-816x.2001.01697.x
36. de Béthune M-P. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: a review of the last 20 years (1989–2009). Antiviral Res. 2010;85(1):75–90. doi:10.1016/j.antiviral.2009.09.008
37. Sluis-Cremer N, Arion D, Parniak MA. Molecular mechanisms of HIV-1 resistance to nucleoside reverse transcriptase inhibitors (NRTIs). Cell Mol Life Sci C. 2000;57(10):1408–1422. doi:10.1007/PL00000626
38. Roers A, Hiller B, Hornung V. Recognition of endogenous nucleic acids by the innate immune system. Immunity. 2016;44(4):739–754. doi:10.1016/j.immuni.2016.04.002
39. Kanneganti T-D. Central roles of NLRs and inflammasomes in viral infection. Nat Rev Immunol. 2010;10(10):688–698. doi:10.1038/nri2851
40. Van Regenmortel MHV. Applying the species concept to plant viruses. Arch. Virol. 1989;104(1):1–17. doi:10.1007/BF01313804
41. Crow YJ. Type I interferonopathies: Mendelian type I interferon up-regulation. Curr Opin Immunol. 2015;32:7–12. doi:10.1016/j.coi.2014.10.005
42. Weidenmaier C, Peschel A. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat Rev Microbiol. 2008;6(4):276–287. doi:10.1038/nrmicro1861
43. Jana B, Chatterjee A, Roy D, et al. Chitosan/benzyloxy-benzaldehyde modified ZnO nano template having optimized and distinct antiviral potency to human cytomegalovirus. Carbohydr Polym. 2022;278(2021):118965. doi:10.1016/j.carbpol.2021.118965
44. Galante AJ, Yates KA, Romanowski EG, Shanks RMQ, Leu PW. Coal-Derived Functionalized Nano-Graphene Oxide for Bleach Washable, Durable Antiviral Fabric Coatings. ACS Appl Nano Mater. 2022;5(1):718–728. doi:10.1021/acsanm.1c03448
45. Alhakamy NA, Ahmed OAA, Ibrahim TS, et al. Evaluation of the antiviral activity of sitagliptin-glatiramer acetate nano-conjugates against sars-cov-2 virus. Pharmaceuticals. 2021;14(3):1–16. doi:10.3390/ph14030178
46. Zakaria MY, Abd El-Halim SM, Beshay BY, Zaki I, Abourehab MAS. ‘Poly phenolic phytoceutical loaded nano-bilosomes for enhanced caco-2 cell permeability and SARS-CoV 2 antiviral activity’: in-vitro and insilico studies. Drug Deliv. 2023;30(1). doi:10.1080/10717544.2022.2162157
47. Dolatyari M, Rostami A. Strong anti-viral nano biocide based on Ag/ZnO modified by amodiaquine as an antibacterial and antiviral composite. Sci Rep. 2022;12(1):1–11. doi:10.1038/s41598-022-24540-8
48. Ullah F, et al. Synthesis and surface modification of chitosan built nanohydrogel with antiviral and antimicrobial agent for controlled drug delivery. Biointerface Res Appl Chem. 2019;9(6):4439–4445. doi:10.33263/BRIAC96.439445
49. Bhattacharya I, Yadavalli T, Wu D, Shukla D. Plasma Membrane-Derived Liposomes Exhibit Robust Antiviral Activity against HSV-1. Viruses. 2022;14(4):1–13. doi:10.3390/v14040799
50. Zakaria MY, Fayad E, Althobaiti F, Zaki I, Abu Almaaty AH. Statistical optimization of bile salt deployed nanovesicles as a potential platform for oral delivery of piperine: accentuated antiviral and anti-inflammatory activity in MERS-CoV challenged mice. Drug Deliv. 2021;28(1):1150–1165. doi:10.1080/10717544.2021.1934190
51. Wang LY, Zhao M-Y, Bu F-Z, et al. Cocrystallization of Amantadine Hydrochloride with Resveratrol: the First Drug-Nutraceutical Cocrystal Displaying Synergistic Antiviral Activity. Cryst Growth Des. 2021;21(5):2763–2776. doi:10.1021/acs.cgd.0c01673
52. Yadav D, Savjani J, Savjani K, Kumar A, Patel S. Pharmaceutical Co-crystal of Antiviral Agent Efavirenz with Nicotinamide for the Enhancement of Solubility, Physicochemical Stability, and Oral Bioavailability. AAPS Pharm Sci Tech. 2022;24(1):7. doi:10.1208/s12249-022-02467-7
53. Damian F, Blaton N, Kinget R, Van Den Mooter G. Physical stability of solid dispersions of the antiviral agent UC-781 with PEG 6000, Gelucire® 44/14 and PVP K30. Int J Pharm. 2002;244(1–2):87–98. doi:10.1016/S0378-5173(02)00316-2
54. Damian F, Blaton N, Naesens L, et al. Physicochemical characterization of solid dispersions of the antiviral agent UC-781 with polyethylene glycol 6000 and Gelucire 44/14. Eur J Pharm Sci. 2000;10(4):311–322. doi:10.1016/S0928-0987(00)00084-1
55. Wang L-Y, Bu F-Z, Yu Y-M, et al. A novel crystalline molecular salt of sulfamethoxazole and amantadine hybridizing antiviral-antibacterial dual drugs with optimal in vitro/vivo pharmaceutical properties. Eur J Pharm Sci. 2021;163:105883. doi:10.1016/j.ejps.2021.105883
56. Yoon J, Kim J, Lee J, et al. Fabrication of antiviral nanofibers containing various Cu salts and ZnO nanorods by electrospinning. J Ind Eng Chem. 2022;116:572–580. doi:10.1016/j.jiec.2022.09.045
57. Chen S, Gao Y, Lou X, et al. Overcoming Bioavailability Challenges of Dasabuvir and Enabling a Triple-Combination Direct-Acting Antiviral HCV Regimen through a Salt of Very Weak Acid for Oral Delivery. Mol Pharm. 2022;19(7):2367–2379. doi:10.1021/acs.molpharmaceut.2c00161
58. Vorozhtsov NO, Yarovaya OI, Roznyatovskii VA, et al. Synthesis and antiviral activity of novel 3-substituted pyrazolinium salts. Chem Heterocycl Compd. 2021;57(4):432–441. doi:10.1007/s10593-021-02921-7
59. Martínez-Gualda B, Sun L, Martí-Marí O, et al. Modifications in the branched arms of a class of dual inhibitors of HIV and EV71 replication expand their antiviral spectrum. Antiviral Res. 2019;168:210–214. doi:10.1016/j.antiviral.2019.06.006
60. Chuchkov K, Chayrov R, Hinkov A, Todorov D, Shishkova K, Stankova IG. Modifications on the heterocyclic base of ganciclovir, penciclovir, Acyclovir - syntheses and antiviral properties. Nucleosides Nucleotides Nucleic Acids. 2020;39(7):979–990. doi:10.1080/15257770.2020.1725043
61. Whittle E, Martín-Illana A, Cazorla-Luna R, et al. Silane modification of mesoporous materials for the optimization of antiviral drug adsorption and release capabilities in vaginal media. Pharmaceutics. 2021;13(9):1416. doi:10.3390/pharmaceutics13091416
62. Qiao Z, Wei N, Jin L, et al. The Mpro structure-based modifications of ebselen derivatives for improved antiviral activity against SARS-CoV-2 virus. Bioorg Chem. 2021;117:105455. doi:10.1016/j.bioorg.2021.105455
63. Spagnoli G, Pouyanfard S, Cavazzini D, et al. Broadly neutralizing antiviral responses induced by a single-molecule HPV vaccine based on thermostable thioredoxin-L2 multiepitope nanoparticles. Sci Rep. 2017;7(1):1–13. doi:10.1038/s41598-017-18177-1
64. Smith BJ, McKimm-Breshkin JL, McDonald M, Fernley RT, Varghese JN, Colman PM. Structural studies of the resistance of influenza virus neuramindase to inhibitors. J Med Chem. 2002;45(11):2207–2212. doi:10.1021/jm010528u
65. Gubareva LV. Molecular mechanisms of influenza virus resistance to neuraminidase inhibitors. Virus Res. 2004;103(1–2):199–203. doi:10.1016/j.virusres.2004.02.034
66. Bantia S, Arnold CS, Parker CD, Upshaw R, Chand P. Anti-influenza virus activity of peramivir in mice with single intramuscular injection. Antiviral Res. 2006;69(1):39–45. doi:10.1016/j.antiviral.2005.10.002
67. Ishizuka H, Yoshiba S, Okabe H, Yoshihara K. Clinical pharmacokinetics of laninamivir, a novel long‐acting neuraminidase inhibitor, after single and multiple inhaled doses of its prodrug, CS‐8958, in healthy male volunteers. J Clin Pharmacol. 2010;50(11):1319–1329. doi:10.1177/0091270009356297
68. Grabrucker AM, et al. Nanoparticles as blood–brain barrier permeable CNS targeted drug delivery systems. In: The Blood Brain Barrier (BBB). Springer; 2013:71–89.
69. Nel A, Ruoslahti E, Meng H. New insights into ‘permeability’ as in the enhanced permeability and retention effect of cancer nanotherapeutics Acs Nano. Vol. 11(10):ACS Publications:9567–9569. 2017
70. Meijer DKF, Jansen RW, Molema G. Drug targeting systems for antiviral agents: options and limitations. Antiviral Res. 1992;18(3–4):215–258. doi:10.1016/0166-3542(92)90058-D
71. Sinha B, Müller RH, Möschwitzer JP. Bottom-up approaches for preparing drug nanocrystals: formulations and factors affecting particle size. Int J Pharm. 2013;453(1):126–141. doi:10.1016/j.ijpharm.2013.01.019
72. Alzaid A, et al. Saudi Parents’ Knowledge, Attitudes, and Practices Regarding Antibiotic use for Upper Respiratory Tract Infections in Children. Int J Pharm Res Allied Sci. 2020;9(1):56.
73. Sankarganesh M, Adwin Jose P, Dhaveethu Raja J, et al. New pyrimidine based ligand capped gold and platinum nano particles: synthesis, characterization, antimicrobial, antioxidant, DNA interaction and in vitro anticancer activities. J Photochem Photobiol B: Biol. 2017;176:44–53. doi:10.1016/j.jphotobiol.2017.09.013
74. Chen Z-L, Huang M, Wang X-R, et al. Transferrin-modified liposome promotes α-mangostin to penetrate the blood–brain barrier. Nanomedicine Nanotechnology, Biol Med. 2016;12(2):421–430. doi:10.1016/j.nano.2015.10.021
75. Drozd KV, Manin AN, Voronin AP, Boycov DE, Churakov AV, Perlovich GL. A combined experimental and theoretical study of miconazole salts and cocrystals: crystal structures, DFT computations, formation thermodynamics and solubility improvement. Phys Chem Chem Phys. 2021;23(21):12456–12470. doi:10.1039/D1CP00956G
76. Nechipadappu SK, Reddy IR, Tarafder K, Trivedi DR. Salt/cocrystal of anti-fibrinolytic hemostatic drug tranexamic acid: structural, DFT, and stability study of salt/cocrystal with GRAS molecules. Cryst Growth Des. 2018;19(1):347–361. doi:10.1021/acs.cgd.8b01451
77. Dengale SJ, Grohganz H, Rades T, Löbmann K. Recent advances in co-amorphous drug formulations. Adv Drug Deliv Rev. 2016;100(2016):116–125. doi:10.1016/j.addr.2015.12.009
78. Buldurun K, Turan N, Bursal E, et al. Synthesis, spectroscopic properties, crystal structures, antioxidant activities and enzyme inhibition determination of Co(II) and Fe(II) complexes of Schiff base. Res Chem Intermed. 2020;46(1):283–297. doi:10.1007/s11164-019-03949-3
79. Venkateswarlu K, Kumar MP, Rambabu A, et al. Crystal structure, DNA binding, cleavage, antioxidant and antibacterial studies of Cu(II), Ni(II) and Co(III) complexes with 2-((furan-2-yl)methylimino)methyl)-6-ethoxyphenol Schiff base. J Mol Struct. 2018;1160(Ii):198–207. doi:10.1016/j.molstruc.2018.02.004
80. Li S-D, Li P-W, Yang Z-M, et al. Synthesis and characterization of chitosan quaternary ammonium salt and its application as drug carrier for ribavirin. Drug Deliv. 2014;21(7):548–552. doi:10.3109/10717544.2013.853708
81. Moser D, Duan Y, Wang F, Ma Y, O’Neill MJ, Cornella J. Selective functionalization of aminoheterocycles by a pyrylium salt. Angew Chem Int Ed. 2018;57(34):11035–11039. doi:10.1002/anie.201806271
82. Alatas F. Perbaikan Kelarutan Albendazol Melalui Pembentukan Kristal Multikomponen dengan Asam Malat. J Farm Galen. 2020;6(1):114–123.
83. Hamill RJ, Sobel J, El‐Sadr W, et al. Comparison of 2 doses of liposomal amphotericin B and conventional amphotericin B deoxycholate for treatment of AIDS-associated acute cryptococcal meningitis: a randomized, double-blind clinical trial of efficacy and safety. Clin Infect Dis. 2010;51(2):225–232. doi:10.1086/653606
84. Budiman A, Citraloka Z Ganesya, Muchtaridi M, Sriwidodo S, Aulifa D Lia and Rusdin A. (2022). Inhibition of Crystal Nucleation and Growth in Aqueous Drug Solutions: Impact of Different Polymers on the Supersaturation Profiles of Amorphous Drugs—The Case of Alpha-Mangostin. Pharmaceutics, 14(11), 2386 10.3390/pharmaceutics14112386
85. Begines B, Ortiz T, Pérez-Aranda M, et al. Polymeric nanoparticles for drug delivery: recent developments and future prospects. Nanomaterials. 2020;10(7):1403. doi:10.3390/nano10071403
86. Yao Y, Zhou Y, Liu L, et al. Nanoparticle-based drug delivery in cancer therapy and its role in overcoming drug resistance. Front Mol Biosci. 2020;7:193. doi:10.3389/fmolb.2020.00193
87. Yoo J, Park C, Yi G, Lee D, Koo H. Active targeting strategies using biological ligands for nanoparticle drug delivery systems. Cancers. 2019;11(5):640. doi:10.3390/cancers11050640
88. Sur S, Rathore A, Dave V, Reddy KR, Chouhan RS, Sadhu V. Recent developments in functionalized polymer nanoparticles for efficient drug delivery system. Nano-Struct Nano-Objects. 2019;20:100397. doi:10.1016/j.nanoso.2019.100397
89. Filipczak N, Pan J, Yalamarty SSK, Torchilin VP. Recent advancements in liposome technology. Adv Drug Deliv Rev. 2020;156:4–22. doi:10.1016/j.addr.2020.06.022
90. Large DE, Abdelmessih RG, Fink EA, Auguste DT. Liposome composition in drug delivery design, synthesis, characterization, and clinical application. Adv Drug Deliv Rev. 2021;176:113851. doi:10.1016/j.addr.2021.113851
91. Maja L, Željko K, Mateja P. Sustainable technologies for liposome preparation. J Supercritical Fluids. 2020;165:104984. doi:10.1016/j.supflu.2020.104984
92. Huang Y, Kuminek G, Roy L, Cavanagh KL, Yin Q, Rodríguez-Hornedo N. Cocrystal solubility advantage diagrams as a means to control dissolution, supersaturation, and precipitation. Mol Pharm. 2019;16(9):3887–3895. doi:10.1021/acs.molpharmaceut.9b00501
93. Cavanagh KL, Kuminek G, Rodríguez-Hornedo N. Cocrystal solubility advantage and dose/solubility ratio diagrams: a mechanistic approach to selecting additives and controlling dissolution–supersaturation–precipitation behavior. Mol Pharm. 2020;17(11):4286–4301. doi:10.1021/acs.molpharmaceut.0c00713
94. Zhang Y-X, Wang L-Y, Dai J-K, et al. The comparative study of cocrystal/salt in simultaneously improving solubility and permeability of Acetazolamide. J Mol Struct. 2019;1184:225–232. doi:10.1016/j.molstruc.2019.01.090
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.